
|
656010, Алтайский край, город Барнаул,
ул.Эмилии Алексеевой, 53 корпус 1,39 корпус2
Телефон / Факс: +7 (3852) 22-62-77
[email protected]
|
Шарко мари тутаБолезнь Шарко — Мари — Тута — ВикипедияМатериал из Википедии — свободной энциклопедии Болезнь Шарко — Мари — Тута (ШМТ), или наследственная моторно-сенсорная нейропатия (НМСН) — наследственная периферическая нейропатия с хроническим прогрессирующим течением. При этой болезни больные страдают от слабости и атрофии мышц дистальных отделов конечностей, деформации стоп и кистей, у них наблюдается снижение сухожильных рефлексов, изменение походки, потеря чувствительности в конечностях[2]. В основе клинических проявлений болезни лежит поражение двигательных и чувствительных периферических нервных волокон. Болезнь Шарко — Мари — Тута диагностируется с приблизительной частотой 1 на 2500 человек. Первое проявление болезни чаще всего происходит в подростковом возрасте или в раннем взрослом возрасте. Тяжесть симптомов сильно различается даже среди членов одной семьи с этим заболеванием. Болезнь Шарко — Мари — Тута нередко ведёт к ограничению трудоспособности и к инвалидизации, при этом большинство пациентов имеют нормальную продолжительность жизни [3]. Болезнь Шарко — Мари — Тута является генетически крайне неоднородным заболеванием, симптомы этой болезни могут быть вызваны мутациями в более чем двух десятках генов, хотя большая часть заболеваний вызвана мутациями в генах PMP22, MPZ, GJB1 и MFN2[4]. Наследование болезни чаще всего аутосомно-доминантное, однако может быть аутосомно-рецессивным и Х-сцепленным[5]. Заболевание носит имена врачей, впервые его описавших в 1886 году: французских врачей Жана-Мартена Шарко и Пьера Мари, а также англичанина Говарда Тута (англ.)русск.[3]. Основные формы болезни Шарко — Мари — Тута[править | править код]Существуют различные формы болезни Шарко — Мари — Тута. Основные формы имеют обозначения ШМТ1, ШМТ2, ШМТ3, ШМТН4, ШМТ5, ШМТ6, ШМТ-ДП, ШМТ-РП и ШМТХ[4]. Причиной ШМТ1 является нарушение миелиновой оболочки периферических нервов, эта форма называется миелинопатия и имеет несколько типов со сходными симптомами. Первые признаки болезни появляются, как правило, в подростковом возрасте. Пациенты испытывают мышечную слабость в ногах, у них происходит атрофия мышц дистальных отделов нижних конечностей, где позднее слабеет и утрачивается чувствительность. Скорость проведения импульса по срединному нерву снижена и составляет менее 38 м/с. У пациентов выявляется сегментарная демиелинизация и ремиелинизация. При биопсии нервных волокон выявляется гиперплазия шванновских клеток с формированием характерного морфологического признака «луковичные головки».
ru.wikipedia.org Болезнь ШаркоХарактерные черты патологии были описаны в конце 19-го века тремя врачами: французами Жаном-Мартином Шарко, Пьером Мари и англичанином Говардом Генри Тутом. В их честь и появилось название – болезнь Шарко-Мари-Тута. Используемые синонимы – наследственная моторно-сенсорная невропатия, невральная амиотрофия. Связана патология с воздействием на периферические нервы, вследствие чего происходит разрушение миелиновой оболочки или длинных отростков нервов – аксонов. Принято считать, что при этом заболевании не происходит поражение ЦНС. Однако есть данные, свидетельствующие о том, что разрушение затрагивает корешки спинного мозга, проводниковые пути. Вследствие нарушения проводимости нервных волокон, расположенных на периферии, атрофируются мышечные ткани конечностей. Постепенно происходит их замена соединительными и жировыми тканями. Симптомы болезни Шарко-Мари-Тута диагностируются чаще всего у детей и молодых людей от десяти до двадцати лет. ПричиныНервные импульсы передаются по длинным отросткам нейронов — аксонам. Они обернуты миелиновой оболочкой. В ее создании принимают участие олигодендроциты. При патологии Шарко-Мари-Тута мутация происходит в гене MFN2. В зоне его ответственности – выработка митохондрального белка. Мутация ведет к формированию сгущений митохондрий в теле аксона. Вероятно, заболевание вызвано еще и воздействием генов на иммунную систему организма. В результате белки миелинового полотна начинают восприниматься как свойственные патологическим бактериям. Активизируется иммунная система, образуются антитела, они проникают сквозь гемато-энцефалический барьер и поражают белковые компоненты. Вследствие воздействия некоторых генов происходит чрезмерная миелинизация нервных клеток, что также нарушает прохождение нервных импульсов. В патогенезе синдрома Шарко-Мари-Тута, таким образом, выделяют 2 формы:
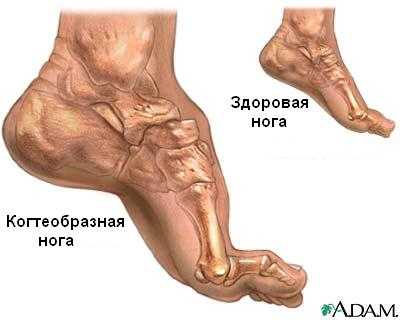
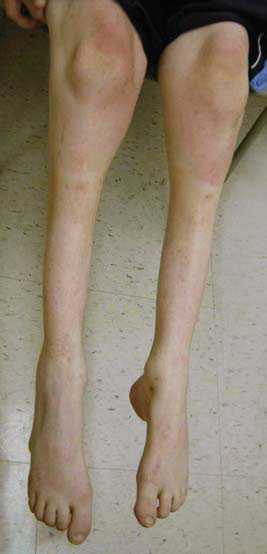
Передача патологии осуществляется преимущественно аутосомно-доминантным путем, т. е. ребенок получает ее от одного из родителей. В некоторых случаях отмечается рецессивная передача – оба родителя являются носителями патологических генов, а для развития болезни нужны две генных копии. Редко возникает мутация гена у одного человека, не связанная с наследственными факторами. Точные причины патологии до настоящего времени неизвестны. СимптомыСиндром Шарко ведет к повреждению двигательных и чувствительных нервов. Разрушение моторных путей сопровождается слабостью и онемением мышечной ткани обеих стоп, быстрым нарастанием усталости. Несколько позже присоединяются болевые ощущения в икроножных мышцах. Развиваются они преимущественно после долгой ходьбы, стояния на одном месте. 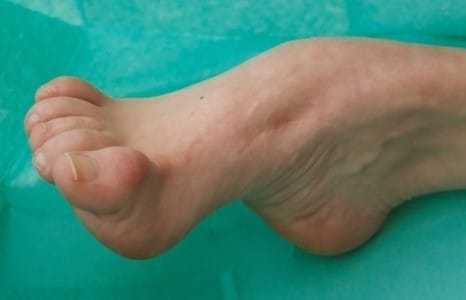 Во время осмотра обнаруживается атрофия мышечных волокон ног. Угнетаются сухожильные рефлексы. Слабость мышц и рефлексов ведет к нарушению походки. Человек падает, покачивается при ходьбе. Атрофия мелких мышечных волокон вызывает изменение формы стопы – свод увеличивается. Наблюдается деформация пальцев. В меньшей степени большого, пальцев, следующих за ним, в большей. Они сгибаются, начинают напоминать когти. Человек теряет способность ходить на пятках. При долгом вынужденном стоянии вынужден переминаться, топтаться, чтобы унять дискомфорт. Поражение икроножной мускулатуры ведет к деформации голени – они начинают напоминать ноги аиста или перевернутой бутылки. Ослабление создает эффект болтающейся стопы. В среднем через 10-15 лет начинается атрофия мускульной ткани рук. Выпадает тонкая моторика. Сначала под удар подпадают дистальные зоны. Кисть начинает напоминать лапу обезьяны. Тело, шея, плечи остаются сохранными. 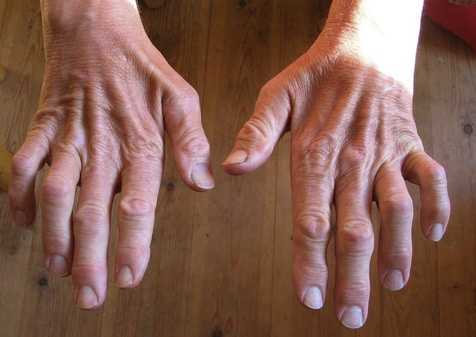 Невральную амиотрофию Шарко-Мари-Тута сопровождают и другие признаки. Среди них:
Если начало заболевания относится к возрасту до 5 лет, у больного со временем нарастают нарушения в работе органов дыхания, внутренних органов, снижается зрение, слух. 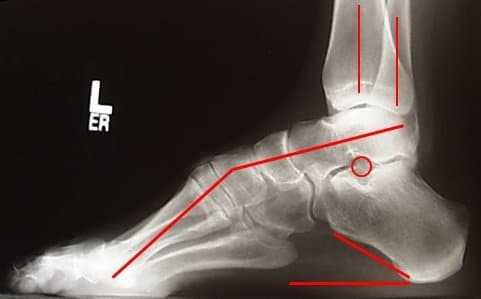 Патология Шарко-Мари-Тута — это медленно прогрессирующая мышечная атрофия. Страдающие от этой формы невропатии долго сохраняют работоспособность. Усиление симптоматики провоцируется травмами позвоночника и головы, инфекционными вирусными и бактериальными заболеваниями. ДиагностикаПодтверждение диагноза синдрома Шарко-Мари-Тута связано, прежде всего, с анализом неврологической симптоматики. Проверяется мускульная сила, сухожильные рефлексы, сенсорная сохранность, тремор. На приеме проверяется деформирование стопы, голени, кисти, позвоночника. Невролог в обязательном порядке проясняет историю появления симптомов, наличие признаков болезни в семье. Назначаются инструментальные методы исследования: электронейрография и электромиография. В первом случае измеряется, с какой скоростью проходят импульсы. Во втором оценивается биоэлектрическая активность мышечных тканей, уточняется степень нарушения периферической системы. Выполняется анализ ДНК. Отрицательный результат еще не подтверждает отсутствия заболевания Шарко-Мари-Тута, т. к. в настоящее время известны не все генетические параметры заболевания. Проведение генетической экспертизы важно при планировании беременности. При невозможности разграничить синдром от других невропатий берется проба тканей мышц и нерва. ЛечениеМетоды терапии патологии Шарко-Мари-Тута связаны с облегчением и уменьшением проявления симптомов. Они включают использование медикаментозных препаратов, физиотерапию и оперативное вмешательство. Медикаментозные препаратыЛекарственная терапия включает средства, направленные на улучшение обмена веществ (Аденозинтрифосфат натрия), микроциркуляции (Пентоксифиллин), нервно-мышечной передачи (Галантамин). Назначается витамин Е, препараты, содержащие кальций. ФизиолечениеПри амиотрофии Шарко-Мари-Тута показано использование методов физиотерапевтического лечения. Применяется массаж, бальнеотерапия, электростимуляция, грязелечение, лечебные ванны, гидромассаж. При симптоматике, связанной с нарушением чувствительности, с осторожностью используют электрофорез и гальванизацию. Если сенсорные нервы не поражены, проводят электрофорез с препаратами кальция и антихолинэстеразы. Основные цели физиотерапевтического лечения болезни Шарко-Мари-Тута:
Хирургическое лечениеОсновная цель операции при патологии Шарко-Мари Тута – предотвратить деформацию стопы. Однако перед проведением операции производится тщательная оценка возможных отрицательных последствий. Анестезия оказывает негативное воздействие на течение болезни. После операции показано ограничение реабилитационных мероприятий. ПрофилактикаАмиотрофию Шарко-Мари-Тута, как и другие генетические заболевания, предупредить невозможно. Однако в силах больного, его родителей, если пациентом является ребенок, облегчить страдания и исключить тяжелые осложнения. Больным рекомендуется носить фиксирующие повязки, которые призваны предотвратить растяжение мышечно-связочных структур. Для поддержания слабеющих лодыжек показано ношение высокой обуви. Прогноз и осложненияНевральная амиотрофия Шарко-Мари-Тута характеризуется медленно прогрессирующим течением. При тяжелом поражении больных ждет потеря способности передвигаться без поддерживающих устройств, колясок. Дисфункция рук сопровождается потерей способности ухаживать за собой. К частым осложнениям относятся вывихи, переломы, растяжения. Болезнь не сокращает продолжительность жизни. Развитие амиотрофии Шарко-Мари-Тута определено наследственными факторами, которые на сегодня еще не до конца изучены. В силу этого возможности подобрать лечение, которое бы остановило его течение и развитие, не существует. Тем не менее, применение симптоматического лечения, физиотерапия, ортопедические устройства позволяют снизить проявление симптомов. neuromed.online Невральная амиотрофия Шарко-Мари-Тута - причины, симптомы, диагностика и лечениеНевральная амиотрофия Шарко-Мари-Тута — прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение. Общие сведенияНевральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания. Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования. Лица мужского пола болеют чаще, чем женщины. По различным данным невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ. Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ. Невральная амиотрофия Шарко-Мари-Тута Патогенетические аспектыНа сегодняшний день неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону. Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути. Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду. КлассификацияВ современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение. Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса, при ШМТ II типа скорость проведения страдает незначительно. Биопсия нерва обнаруживает при I типе сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток; при II типе — дегенерацию аксонов. СимптомыНевральная амиотрофия Шарко-Мари-Тута начинается с развития симметричных мышечных атрофий в дистальных отделах ног. Начальные симптомы манифестируют, как правило, в первой половине второго десятилетия жизни, реже в период от 16 до 30 лет. Они заключаются в повышенной утомляемости стоп при необходимости длительно стоять на одном месте. При этом наблюдается симптом «топтания» - чтобы снять утомляемость стоп пациент прибегает к ходьбе на месте. В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов. Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок. Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса. Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей. Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей. Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет. Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние. Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз. ДиагностикаВозраст начала заболевания, его типичная клиника, симметричный характер поражения, медленное неуклонное распространение атрофий и усугубляющаяся в связи с этим симптоматика во многих случаях позволяют предположить невральную амиотрофию. Проводимый неврологом осмотр выявляет мышечную слабость в стопах и голенях, деформацию стоп, отсутствие или значительное снижение ахилловых и коленных рефлексов, гипестезию стоп. Для дифференциации ШМТ от других нервно-мышечных заболеваний (миотонии, миопатии, БАС, невропатией) проводится электромиография и электронейрография. С целью исключения метаболической невропатии проводится определение сахара крови, исследование гормонов щитовидной железы, тест на наркотики. Всем пациентам для уточнения диагноза рекомендована консультация генетика и проведение ДНК-диагностики. Последняя не дает 100% точного результата, т. к. пока известны не все генетические маркеры ШМТ. Более точным способом диагностики является введенное в 2010г. секвенирование генома. Однако это исследование пока является слишком дорогостоящим для широкого использования. Иногда возникают затруднения в дифференциальной диагностике болезни Шарко-Мари-Тута с невритом Дежерина – Сотта, дистальной миопатией Гоффманна, хроническими полиневропатиями. В таких случаях может потребоваться биопсия мышц и нервов. ЛечениеНа современном этапе радикальные способы лечения генных болезней не разработаны. В связи с этим применяется симптоматическая терапия. Проводятся повторные курсы внутримышечного введения витаминов группы В и витамина Е. С целью улучшения мышечной трофики используют АТФ, инозин, кокарбоксилазу, глюкозу. Назначаются ингибиторы холинэстеразы (неостигмин, оксазил, галантамин), препараты для улучшения микроциркуляции и антиоксиданты (никотиновая кислота, пентоксифиллин, мельдоний). Наряду с фармакотерапией по рекомендации физиотерапевта активно применяются физиотерапевтические методики: электрофорез, СМТ, электростимуляция, диадинамотерапия, грязелечение, УЗ-терапия, оксигенобаротерапия. Целесообразно водолечение сероводородными, сульфидными, хвойными, радоновыми лечебными ваннами. Большое значение в сохранении двигательной активности пациентов, предотвращении развития деформаций и контрактур имеют ЛФК и массаж. При необходимости ортопедом назначается ортопедическое лечение. www.krasotaimedicina.ru Болезнь Шарко Мари Тута - проявление патологииЧаще всего болезнь Шарко Мари Тута передается по наследству и имеет врожденную форму. Заболевание приводит к нарушению чувствительности и изменению подвижности. Если патологию не лечить, то будут осложнения. По началу это может проявляться в виде хромоты, но потом это приведет к нарушению дыхательной системы. Медики говорят, что патология передается по генетике и встречается довольно часто. Если кто-то из родственников имеет заболевание, то большая вероятность, что у их детей будут данные нарушения. Чаще всего невральная амиотрофия выявляется у мужчин. При первых симптомах необходимо обращаться к врачу. СимптомыЗаболевание Шарко Мари Тута может проявляться по-разному и главное вовремя узнать развитие болезни. Это нужно для того чтобы своевременно обратиться в медицинское учреждение. Если пациента беспокоят некоторые признаки, то не рекомендуется игнорировать симптомы и посетить врача. У детей заболевание практически протекает незаметно, поэтому выявить патологию можно только у ребенка старше 14 лет.
Основные симптомы при развитии заболевания:
Невральная амиотрофия может развиваться незаметно и медленно. Если наследственная нейропатия Шарко–Мари–Тута прогрессирует уже пару лет, то у больного может быть повреждение нервных тканей, как рук, так и ног. Перемежающийся дистрофический синдром возникает чаще всего в предплечье и кистях. Мышцы туловища поражаются и со временем происходит их атрофия. Тогда и возникает деформация позвоночника.
Спровоцировать ускорение патологии и ухудшить самочувствие могут инфекции, интоксикация и травмы. Если стали заметны даже небольшие подозрения на невральную амиотрофию, то необходимо не медлить и обращаться к врачу. ДиагностикаЧаще всего невральная амиотрофия начинается еще в подростковом возрасте. Мышцы начинают медленно атрофироваться, и поэтому невропатолог может подозревать Шарко-Мари-Тута. В первую очередь специалист проводит неврологический осмотр. Это помогает увидеть нарушение движений и мышечную атрофию. Поставить точный диагноз получается с трудом, для этого необходимо пройти комплексное обследование. Может назначаться анализ ДНК для того чтобы установить тип наследования. Такие методы исследования точной информации предоставить не смогут, так как генетические маркеры еще не до конца изучены. Применяется также электронейромиография она дает возможность увидеть скорость импульсов, которые проходят по нервным окончаниям.
Специальная подготовка к процедуре не нужна, но перед ней необходимо снять часы и различные украшения. Как правило, больной должен быть в положении полусидя и в кресле при этом все мышцы нужно расслабить. Противопоказаний для проведения ЭМГ практически нет, но нельзя проходить процедуру, если имеются имплантанты и кардиостимуляторы. Специалист часто назначает биопсию мышцы или нерва, это помогает подтвердить точный диагноз. Врач может направить на анализ, который позволяет определить уровень сахара в крови. Назначается исследование на уровень гормонов щитовидной железы. Если возникли неприятных симптомы, то обязательно нужно обратиться к врачу. Если игнорировать признаки невральной амиотрофии то это приведет к тому, что будут серьезные последствия. Народные способы леченияДля того чтобы избавиться от невральной амиотрофии можно применять народные способы лечения. Существует большое количество различных рецептов и готовятся они в домашних условиях. Дает положительный результат в терапии настойка на основе яичной скорлупы, меда, лимона и коньяка. Это поможет избавиться от недостатка кальция и укрепит мышцы. Для начала берется скорлупа от яиц, но она должна быть обязательно белого цвета. Нужно их хорошо вымыть и положить в трехлитровую банку. Взять около десяти лимонов и выжать их в банку и крепко обвязать её марлей.
Поставить в темное место примерно на неделю, всё зависит от того, как быстро растворится скорлупа. После этого туда поместить немного теплого липового меда и влить стакан коньяка. Полученная настойка должна находится в прохладном и темном месте. Принимать внутрь нужно несколько чайных ложек в день после еды. Специальный отвар при невральной амиотрофии готовится из корня аира, кукурузных рылец, спорыша, шалфея, льнянки. Взять около сто грамм каждого растения и залить кипятком в термосе. Поставить на ночь для того чтобы настоялся и утром уже можно принимать. Употреблять необходимо за час до еды около четырех раз в день.
Настойка из овса для того чтобы приготовить средство необходимо взять овсяных зерен. Промыть их тщательно и засыпать в стеклянную банку. В емкость добавить сахара и лимонного сока и залить чистой водой. Настойка будет готова примерно через несколько суток. Употреблять можно несколько раз в день.
Дают положительный результат специальные компрессы, которые делаются из камышовых метелок. Для этого необходимо взять три жмени и залить кипятком на сорок минут. Достать их из жидкости и дать время, чтобы они слегка остыли. Дальше нужно их прикладывать к больным местам и крепить бинтами. Можно укутать сверху теплым шарфом и продержать камышовые метелки пока они не остынут. После того как они будут холодными их необходимо снять и сделать массаж всех конечностей. Хорошо помогает от онемения конечностей настойка на основе чеснока. Необходимо взять стеклянную банку и наполнить её чесночной массой залить это всё водкой. Накрыть крышкой и поставить в темное место на несколько недель, но иногда нужно встряхивать банку. Перед употреблением необходимо около 5 капель разбавлять водой и пить несколько раз в день. Как правило, курс лечения проводится в течение месяца.
При поражении суставов кистей можно попробовать сделать специальные ванночки. Для этого берется кожура моркови, лука, картофеля, свеклы и варится это всё в пятилитровой емкости. Дальше нужно будет слить жидкость в тазик и делать там ванночки. Можно добавить туда немного йода и соли. Нужно делать вовремя процедуры массаж и после окончания надеть теплые перчатки. Если делать всё правильно, то народные рецепты помогут избавить от недуга. Лучше все данные способы сочетать вместе с другими методами лечения.
ЛечениеОсновная причина развития невральной амиотрофии является генетическая предрасположенность. Вылечить полностью заболевание не получится, но замедлить прогрессирование патологии можно. Существует ряд методов, которые помогают устранить симптоматику болезни. Применяются следующие способы терапии:
Если соблюдать все рекомендации врача, то это поможет устранить симптомы в короткие сроки. Главное, вовремя обратиться к врачу для того чтобы он назначил лечение. Если игнорировать первые проявления невральной амиотрофии то это может привести к серьезным последствиям.
Назначаются следующие медикаменты для облегчения состояния больного:
Лечение подбирается под каждого пациента индивидуально. Всё зависит от того на какой стадии развития заболевание. Для того чтобы больной почувствовал себя легче, нужно будет применять определенные методы лечения. Как правило, одних медикаментов бывает недостаточно, поэтому необходимо сочетать с другими способами терапии. Лучше всего обращаться к врачу при первых проявлений патологии. Массажные процедуры хорошо помогают справиться с признаками невральной амиотрофии. Массаж делается, как руками, так и аппаратом и происходит воздействие на тело. Массажные процедуры захватывают кожу, мышцы, сосуды, нервы. Если использовать несколько видов массажей, то это прежде всего поможет расслабиться. Можно добиться тонизирующего эффекта, болеутоляющего и так далее. Важно также соблюдать правильное питание, ведь это играет большую роль в терапии. Невральная амиотрофия приводит к утрате мышечной массе, поэтому придется, чтобы рацион был сбалансированным. Необходимо, чтобы была белковая диета и в питании присутствовали аминокислоты. Может быть так, что организм не усваивает белок. Тогда придется искать причину почему, так происходит. Должны обязательно проводится физические упражнения. Перед тем, как их выполнять врач должен индивидуально подбирать. При заболевании происходит отказ определенных мышц и другие берут на себя работоспособность пораженных участков. Если неправильно будут назначены упражнения, то от лечения не будет никакого эффекта. Только врач должен подбирать под каждого больного определенные физические нагрузки. Взрослым людям часто нужна психологическая помощь при заболевании. Когда происходит развитие патологии, то у них могут возникать нехорошие мысли. На подобии того, что они приносят только лишние проблемы своим родным из-за болезни.
В детском возрасте нужна помощь только в том, случае, если необходимо развиваться интеллектуально. Ведь, как правило, мышцы также учувствуют в развитии головного мозга. Для того чтобы не допустить отставание ребенка нужно применять специальные интеллектуальные упражнения. Существует ряд продуктов, которые запрещается употреблять при заболевании. Отказаться необходимо от алкоголя и энергетиков. Сладкие напитки и кофе также нужно исключить из рациона. Употребление соли необходимо сократить, так как это провоцирует развитие заболевания. Продукты быстрого приготовления и фаст-фуд приводят к разрушению мышечных волокон. Патология называется Шарко-Мари-Тута или наследственная моторно-сенсорная нейропатия. ОсложненияЕсли запустить невральную амиотрофию, то могут быть необратимые последствия. Проявляются они в виде нарушения дыхательной системы. Если патология поражает нервные окончания, которые контролируют диафрагму. Больному скорее всего нужно будет употреблять бронхолитические средства или искусственную вентиляцию легких. Лишний вес или сильное ожирение может привести к тому, что пациенту становится трудно дышать. Депрессивное состояние может быть из-за стрессовых ситуаций. Чаще всего возникает это из-за прогрессирования патологии. Могут применяться, как специальные лекарственные препараты, так и психологическая помощь. Если заболевание будет в запущенной форме и без лечения, то это приведет к инвалидности.
Пациент может перестать полностью передвигаться и на этой фоне возникнуть глухота. Для того чтобы не было такие серьезных последствий, необходимо своевременно обращаться к врачу. Он сможет назначить комплексную диагностику, чтобы поставить точный диагноз. Только после этого будут применяться методы лечения. Заболевание предоставляет хроническую моторную и сенсорную полинейропатию. Что делать, если обнаружено заболеваниеЛечение помогает улучшить состояние и замедлить развитие невральной амиотрафии. Но всё равно необходимо будет придерживаться некоторых правил и вести определенный образ жизни. Нужно будет почаще делать физические нагрузки. Если сразу же начать их выполнять, то это быстрее принесет положительный эффект. Человек, который болеет данным заболеванием, ему необходимо выбрать такой вид спорта, чтобы были незначительные нагрузки. Это может быть велосипед, плавание, катание на лыжах. Работа также должна быть специально подобрана под пациента и без сильных физических напряжений. Обувь обязательно должна быть удобной и не давить. Если у больного уже имеется «свисающая стопа», то ему нужно будет подбирать себе туфли под заказ, чтобы там был специальный фиксирующий ортез. Если обувь будет правильная, то это поможет избежать падения и травмы. Обязательно питание должно быть правильным, это дает возможность избежать лишних килограмм. Ведь ожирение приносит дополнительную нагрузку на слабые мышцы. В рационе должны быть антиоксиданты и витамины группы А,С,Е. Стараться не допускать простуды и переохлаждения, это поможет чувствовать себя лучше.
На сегодняшний день медики не могут полностью устранить невральную амиотрофию. Если получилось так, что болезнь затронула пациента, то не стоит переживать. Нужно будет слегка поменять свой образ жизни. Чем раньше обратиться к врачу, тем быстрее можно избавиться от симптомов заболевания. Лечение должно быть назначено только квалификационным врачом. Заниматься самолечением не стоит, иначе последствий не избежать. При первых симптомах лучше всего посетить сразу же медицинское учреждение. Врач направит на определенные исследования и только после этого назначит комплексную терапию. Если невральная амиотрофия будет запущенно, то серьезных последствий не избежать. nevrology.net Факты о наследственной мото-сенсорной нейропатии Шарко-Мари-ТусПо материалам Американской Ассоциации Мышечных дистрофий ЧТО ТАКОЕ — ЗАБОЛЕВАНИЕ ШАРКО-МАРИ-ТУС? Заболевание Шарко-Мари-Тус (ШМТ) – это неврологическое заболевание, названное так по именам трех врачей, впервые описавших его в 1886 году: Jean-Martin Charcot и Pierre Marie из Франции, и Howard Henry Tooth из Великобритании В отличие от других неврологических заболеваний, ШМТ обычно не является жизнеугрожающим, и почти никогда не поражает головной мозг. При ШМТ поражаются периферические нервы – участки волокон нервных клеток, соединяющие головной и спинной мозг с мышцами и органами чувств Периферические нервы контролируют движения посредством передачи импульсов от спинного мозга к мышцам. Они передают ощущения путем путем проведения сигналов, таких как боль или температура, от рук и ног к спинному мозгу. Они также помогают контролировать баланс посредством передачи информации о положении тела в пространстве Они передают информацию о руках и ногах в спинной, и далее – в головной мозг, таким образом мозг «узнает», где находится нога при ходьбе, или где должна находиться рука, чтобы до чего-то достать. Повреждение нервов, или нейропатия, приводит к слабости и истощению мышц, а также некоторой потере чувствительности, особенно – в удаленных участках тела: стопах, голенях, кистях рук и предплечьях Хотя ШМТ может выглядеть очень похоже на приобретенные нейропатии – тип нервных повреждений, вызванных диабетом, иммунными аномалиями, или воздействием определенных химикатов или лекарств – она не вызвана какими то действиями человека, и не является инфекционной. ШМТ – заболевание наследственное, что означает, что оно может передаваться в семье от поколения к поколению (См. «Это семейное?») Исходя из всех этих особенностей ШМТ иногда называют «наследственной мото-сенсорной нейропатией» (НМСН). Некоторые врачи также используют устаревшее название «перонеальная мышечная атрофия», связанное с истощением перонеальных мышц голени Существует даже больше названий для ШМТ, поскольку заболевание представлено многими различными формами, различающиеся по тяжести течения, возрасту начала, прогрессии и специфическим симптомам. Например, заболевание Дежерина-Сотта (ДС) – тяжелая форма ШМТ, дебютирующая в младенчестве или раннем детстве Хотя не существует радикального лечения ШМТ, есть терапевтические методы, которые можно использовать для эффективного воздействия на ее симптомы. Такие методы, описанные в этой статье вместе с общим обзором ШМТ, дают возможность многим людям с этим заболеванием вести активную и продуктивную жизнь ЧЕМ ВЫЗВАНА ШМТ? ШМТ обусловлена дефектами генов, являющихся сегментами ДНК, содержащейся в хромосомах клеток тела. Гены – это «рецепты» для синтеза протеинов, выполняющих важные функции в нашем организме. Каждая форма ШМТ связана со специфическим геном, и все эти гены ответственны за синтез протеинов, расположенных в периферических нервах.
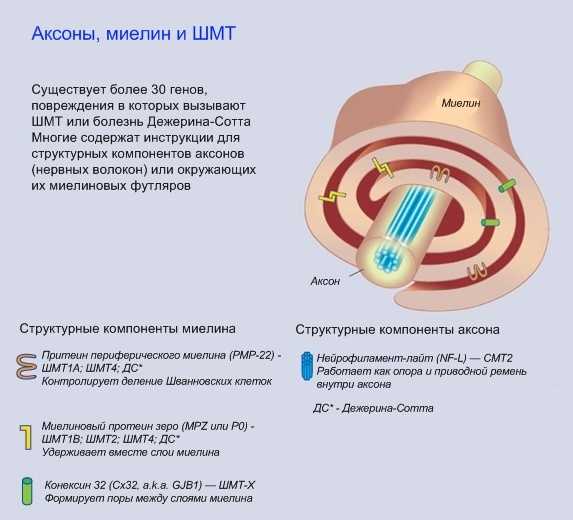
Периферические нервы осуществляют важную связь между головным мозгом и остальным телом. Когда вы решаете пошевелить ногой, ваш головной мозг посылает электрические сигналы контролирующим мышцы нервным клеткам в спинном мозге, которые затем используют периферические нервы для передачи сигналов мышцам ноги. А если вы ушибете ногу, вы почувствуете это, поскольку чувствительные к боли нервные клетки пошлют сигнал по периферическим нервам в головной мозг. Периферические нервы являются частью волокон, или аксонов, которые тянутся от чувствительных или контролирующих мышцы нервных клеток, и проводят сигналы от спинного мозга или к нему Для того, чтобы ваши движения и реакции были быстрыми и точными, аксоны должны передавать сигналы за доли секунд. Это серьезный вызов для аксонов, которые протянуты на длинные расстояния, как например те, которые соединены с мышцами пальцев рук и ног. Для повышения производительности каждый аксон окружен оболочкой, называемой миелином. Подобно тому, как пластиковая оболочка используется для изоляции электрических проводов, миелин изолирует электрические сигналы в аксонах. Он также дает аксонам необходимое питание. Примерно 20 генов затрагиваются при ШМТ, каждый из которых связан с определенным типом (а во многих случаях – с несколькими типами) этого заболевания (см. «Каковы различные типы ШМТ?») Некоторые из этих генов отвечают за синтез протеинов, необходимых для аксонов, другие же – за синтез протеинов, необходимых для миелина Поврежденные миелиновые гены могут вызвать разрывы миелина (называемые демиелинизацией), в то время как поврежденные гены аксонов могут привести к ухудшению функций аксонов (аксонопатия) В любом случае конечный результат таков: дефекты аксонов или миелина вызывают прогрессирующее повреждение аксонов Наиболее протяженные аксоны тела особенно чувствительны к повреждениям, что объясняет, почему при ШМТ наибольшие моторные и сенсорные проблемы возникают в конечностях При наиболее тяжелых формах ШМТ могут быть повреждены и другие нервы, а не только те, которые идут к конечностям и от них. Если повреждены нервы, идущие к диафрагме и межреберным мышцам и от них, это может привести к проблемам с дыханием ЧТО ПРОИСХОДИТ С ЧЕЛОВЕКОМ, ИМЕЮЩИМ ШМT, И КАК ЕЕ ЛЕЧИТЬ? Отчасти потому, что существуют различные типы ШМT, точные симптомы значительно варьируются от человека к человеку. Этот раздел содержит общую картину ШМT, а в следующем разделе описаны различные виды заболеваний. Мышечная слабость В основном, люди с ШМT испытывают медленно прогрессирующую слабость и истощение дистальных мышц, контролирующих конечности. Эти мышцы контролируют движения стоп и кистей. Большинство проксимальных мышц, т.е. тех, что расположены ближе к туловищу, таких, как мышцы рук и ног, затрагиваются реже. Обычно слабость начинается в стопах и голенях, проявляясь «свисающей стопой» — сложностями с подтягиванием стопы к голени, так что пальцы при ходьбе расположены очень низко. «Свисающая стопа» приводит к частым спотыканиям, и по мере развития слабости и попытке скомпенсировать ее больные вырабатывают аномальную походкуь ть вначале проявляется в стопах и голенях Многие люди с ШМТ впервые обращаются к неврологу после того, как становятся частыми обусловленные «свисающей стопой» спотыкания, падения, растяжения или переломы голеностопа Когда подобные проблемы возникают, некоторые люди находят, что могут преодолевать их просто ношением высокой обуви, поддерживающей голень Другим могут потребоваться фиксаторы на ногах, такие как голеностопные ортезы (AFO) — съемные формочки, плотно подогнанные по стопе и голени. Некогда представлявшие из себя тяжелые металлические конструкции, требующие ношения специальной обуви, теперь AFO изготавливаются из легкого пластика, индивидуально по форме ног, и могут носиться под брюками и легкими туфлями Для людей с большей проксимальной слабостью существуют коленоголеностопные ортезы (KAFO), которые простираются по ноге чуть выше колена. Обычно их можно носить под брюками. Некоторые ортезы дают возможность двигаться голени или колену, в то время как другие предотвращают движения для осуществления большей поддержки Большинству людей с ШМТ не требуется кресло-коляска или мото-скутер, однако пожилым людям с развитой стадией ШМТ или людям с тяжелым типом заболевания может потребоваться для передвижения какое-то из этих устройств, особенно когда нужно преодолеть большое расстояние. Простыми креслами-колясками, как и AFO, эти люди не могут воспользоваться. Но существуют кресла-коляски, которые можно использовать практически на любой местности — от торговых центров до походных маршрутов, многие из которых управляются простым переключателем По мере развития заболевания многие люди испытывают слабость в мышцах кистей и предплечий, что вызывает трудности с выполнением действий, требующих точных движений пальцев, таких как поворот дверных ручек, застегивание пуговиц или «молний» на одежде. Зачастую эти проблемы могут быть преодолены с помощью трудотерапии, которая помогает людям выполнять повседневные действия с использованием различных приспособлений Например, трудотерпаевт может рекомендовать укрепить на дверях специальные прорезиненные ручки, или использовать одежду на кнопках или «липучках» Слабость дыхательных мышц при ШМТ встречается редко, однако если это происходит — это может быть опасным для жизни. Если вы регулярно испытываете одышку, необходимо проверить дыхание у специалиста, который может посоветовать вам периодическое применение или применение в ночное время приборов, подающих воздух в легкие под давлением Некоторые люди с ШМТ испытывают тремор (непроизвольное дрожание), хотя как правило это не вызывает серьезной нетрудоспособности или дискомфорта. Вариант ШМТ с выраженным тремором иногда называют синдромом Русси-Леви Контрактуры и деформации костей У многих людей с ШМТ в конце концов развиваются контрактуры (малоподвижность суставов), что приводит к деформациям стоп и кистей рук Контрактуры развиваются из-за того, что некоторые мышцы около сустава ослаблены, а другие сохраняют свою силу, сокращаясь и растягивая сустав. Со временем кости в районе сустава смещаются, занимая неправильное положение Например, если мышцы, поднимающие стопу к голени, ослабевают, а мышцы на нижней поверхности стопы сокращены и натянуты, это приводит к наиболее распространенному типу деформации стопы — укороченной стопе с высоким сводом (pes cavus) По мере того, как контрактура ухудшается, пальцы фиксируются в изогнутом положении У некоторого количества людей с ШМТ напротив формируется плоскостопие (pes planus), предположительно из-за другого типа мышечной слабости При ходьбе эти деформации могут вызывать нехарактерное трение в районе пальцев, пятки и подошвы, приводя к образованию болезненных ссадин, волдырей и мозолей. Если не лечить, то контрактуры и вторичные повреждения стоп прогрессируют, вызывая все большие трудности при ходьбе По мере прогрессирования ШМТ контрактуры в кистях рук могут зафиксировать пальцы в согнутом положении, а в редких случаях тяжелая проксимальная слабость может вызвать сколиоз (поперечное искривление позвоночника) или кифоз (продольное искривление) У небольшого числа людей с тяжелой формой ШМТ в раннем возрасте развивается смещение бедер Одним из наиболее эффективных способов уберечь мышцы от перенапряжения и формирования контрактур является регулярная программа физиотерапии, которая обычно включает умеренные упражнения и растяжки Развитие контрактур стоп можно также замедлить путем применения AFO, которые удерживают стопы в нормальной позиции и уменьшают давление на голени. Аналогично фиксация может быть использована для предотвращения непроизвольного сгибания пальцев ног и рук Если эти методы не помогли и развились серьезные контрактуры, возможно применение хирургических методов для освобождение зажатых мышц и суставов или корректировки деформаций костей. Хирургическое вмешательство зачастую необходимо и при развитом сколиозе Потеря чувствительности и связанные с ней симптомы Поскольку при ШМТ поражаются чувствительные аксоны, большинство людей с ШМТ имеют пониженную чувствительность к горячему, к боли и прикосновениям в стопах и нижней части ног Хотя пациенты с ШМТ зачастую жалуются, что стопы становятся холодными (что вызвано как истощением изолирующих мышц, так и повреждением чувствительных аксонов) большинство из этих проблем с чувствительностью можно выявить только при неврологическом осмотре, но важно признать, что они существуют В сочетании с регулярными ссадинами вследствие деформации стоп, потеря болевой чувствительности при ШМТ повышает риск образования язвочек — ран, остающихся незамеченными, и поэтому серьезно инфицированными. Если у вас ШМТ, и особенно если у вас деформированы стопы, вам необходимо регулярно осматривать стопы на предмет повреждений Как ни парадоксально, но некоторые пациенты с ШМТ испытывают бОльшую боль — комбинацию болезненных мышечных судорог и нейропатической боли. Эта боль вызвана не внешними воздействиями, а дефектными сигналами в чувствительных аксонах. Оба типа боли обычно можно купировать медикаментозно У многих людей с ШМТ потеря чувствительности ассоциируется с сухостью кожи и выпадением волос в пораженной области В редких случаях потеря чувствительности может вызывать постепенное ухудшение слуха, а иногда — и глухоту. Зная об этих потенциальных проблемах, можно вовремя начать лечение в случае необходимости Опасность приема некоторых препаратов Применение определенных предписанных препаратов или злоупотребление алкоголем может вызвать приобретенную нейропатию, и таким образом усугубить течение ШМТ. Изучение различных случаев показало, что применяемый при химиотерапии препарат винкристин может вызвать резкое ухудшение у людей с ШМТ Когда вам впервые назначается какой-либо препарат, неплохо проконсультироваться со своим лечащим врачом о его его возможных эффектах при ШМТ. Или по крайней мере воспользоваться интернетом, чтобы внимательно ознакомиться с инструкцией по применению конкретного препарата, чтобы лучше понять его действие и возможные побочные реакции Вряд ли там будет что-то специфическое, касающееся именно ШМТ. Однако если в описании побочных эффектов встречаются слова «нейропатия», «парестезия», «нейропатическая боль» или «повреждение периферических нервов», вам следует проконсультироваться с лечащим врачом о применении этого препарата при ШМТ и возможных альтернативах Список препаратов, противопоказанных при ШМТ, часто состоит в основном из тех, которые применяются для лечения серьезных заболеваний, таких, как рак. В этом случае возможно и не найдется альтернативы, поэтому необходимо осознавать, что симптомы ШМТ могут ухудшиться Примечание
Потенциально токсичные при ШМТ препараты
Источник: Charcot-Marie-Tooth Neuropathy GeneReview Имеющие явно выраженный высокий риск Vinca alkaloids (Vincristine) Имеющие риск от умеренного до значительного Amiodarone (Cordarone) Имеющие неопределенный или незначительный риск 5-Fluoracil Имеющие незначительный или сомнительный риск Allopurinol КАКОВЫ РАЗЛИЧНЫЕ ТИПЫ ШМТ? Многочисленные типы ШМТ различаются по возрасту начала, типу наследования, тяжести течения и тем, что в первую очередь поражено — аксоны или миелин Хотя эти различия и используются на практике, важно понимать, что вследствие значительного числа генетических дефектов, приводящих к развитию ШМТ, некоторые люди попадают на границу между различными типами и имеют специфические «подтипы», не описанные в этой статье Подробнее о генетике и наследовании ШМТ см. «Это семейное?» ШМТ1 и ШМТ2 Начало: обычно детство или юность Наследование: тип 1 — аутосомно-доминантное; тип 2 — аутосомно-доминантное или аутосомно-рецессивное Особенности: Оба эти типа — наиболее распространенные формы ШМТ (фактически подтип ШМТ1А, обусловленный дефектом гена PMP22 на хромосоме 17 составляет примерно 60% всех случаев ШМТ) ШМТ1 вызвана демиелинизацией, а ШМТ2 — аксонопатией, но оба типа имеют классические симптомы, описанные выше ШМТ2 иногда ассоциирована с поддающимся лечению состоянием, именуемым «синдромом беспокойный ног» — непреодолимыми позывами к движениям ногами в положении сидя или лежа ШМТ-Х Начало: детство или юность Наследование: Х-сцепленное Особенности: Симптомы ШМТ-Х схожи с таковыми при ШМТ1 и ШМТ2. Но поскольку связана с Х-хромосомой, у мальчиков как правило более тяжелое течение, чем у девочек ШМТ4 Начало: младенчество, детство или юность Наследование: аутосомно-рецессивное Особенности: ШМТ4 — демиелинизирующая форма ШМТ, вызывающая в основном дистальную слабость, но иногда вовлечены и проксимальные мышцы. Также наблюдаются расстройства чувствительности. В случае начала в младенчестве ШМТ4 характеризуется сниженным мышечным тонусом. У маленьких детей с ШМТ4 обычно замедленное моторное (связанное с движением) развитие Болезнь Дежерина-Сотта (ДС) Начало: раннее детство (обычно до 3 лет) Наследование: аутосомно-доминантное или аутосомно-рецессивное Особенности: ДС иногда классифицируется как подгруппа ШМТ4, а также иногда обозначается как НМСН3. Это тяжелая нейропатия с генерализованной слабостью, иногда приводящей к тяжелой инвалидизации, потере чувствительности, искривлению позвоночника, и иногда — легкой потере слуха В основе ДС мутации в тех же генах, что и при других различных формах ШМТ Конгенитальная (врожденная) гипомиелинизированная нейропатия (КГН) Начало: врожденная (или сразу после рождения) Наследование: аутосомно-рецессивное, спонтанное Особенности: В отличие от других типов ШМТ, КГН ассоциирована со сниженным формированием миелина с рождения, а не с разрушением имеющегося миелина. Генетически и клинически схожа с ДС, но обычно с более ранним началом и с непрогрессирующим или медленно прогрессирующим течением Многие дети с КГМ взрослеют и у них происходит постепенное увеличение силы КАК ШМТ ДИАГНОСТИРУЕТСЯ? Комбинация слабости голеней и деформаций стоп — «красный флаг» для ШМТ, но все же этого недостаточно для диагнозa. Когда у пациента есть эти симптомы, грамотный невролог обычно начинает с физикального обследования для выявления дополнительных признаков дистальной слабости и утраты чувствительности В качестве теста слабости ног невролог может попросить пациента походить на пятках, либо попытаться двигать различными участками ноги с преодолением усилия. Для выявления потери чувствительности неврологи обычно используют исследование глубоких сухожильных рефлексов (например коленный рефлекс), которые ослаблены либо отсутствуют у людей с ШМТ В процессе первоначальной оценки невролог также спросит пациента о семейной истории. Семейная история ШМТ-подобных симптомов в сочетании с признаками повреждения нервов при индивидуальном физикальном обследовании решительно указывают на ШМТ или другую наследственную нейропатию Отсутствие семейной истории не исключает ШМТ, но в этом случае невролог может спросить о диабете, чрезмерном воздействии определенных препаратов и других потенциальных причинах нейропатии Далее, если в качестве диагноза все еще предполагается ШМТ, невролог может назначить генетическое исследование. Эти тесты, проводимые на образцах крови, разработаны для выявления наиболее частых генетических дефектов, известных как причина ШМТ. По ДНК в образцах крови можно выявить многие, хотя конечно и не все, генетические мутации, лежащие в основе ШМТ Положительные результаты генетического теста дают определенный диагноз и информацию, которую можно использовать для планирования семьи. Но следует подчеркнуть еще раз: негативный результат не опровергает ШМТ. Невролог также может провести исследование скорости нервной проводимости (СНП), в процессе которого измеряется сила и скорость электрических сигналов, проходящих по нервам Тест заключается в размещении поверхностных электродов, аналогичных тем, что применяются для электрокардиограммы, на кожу в различных точках вблизи нервов. Один из электродов производит импульс, стимулирующий электрических ответ в нерве, другой фиксирует этот ответ, прошедший по нерву Замедленный ответ является признаком демиелинизации, а слабый ответ — признаком аксонопатии. Таким образом, СНП часто используется для дифференцирования ШМТ1 и ШМТ2 Другие процедуры, иногда применяемая в диагностике ШМТ — это электромиография (ЭМГ), в процессе которой измеряются электрические сигналы в мышцах, и, реже, биопсия нерва, заключающаяся в изъятии и последующем исследовании небольшого образца нерва ЭТО СЕМЕЙНОЕ? ШМТ может проявиться в семье, даже если нет явной семейной истории заболевания. В частности, это может произойти потому, что ШМТ может наследоваться тремя разными путями, что не всегда легко проследить по родословной: Х-сцепленному, аутосомно-доминантному или аутосомно-рецессивному Х-сцепленное наследование означает, что генетический дефект (или мутация) локализован на Х-хромосоме. У женщин, имеющих две Х-хромосомы, нормальная копия гена на одной хромосоме зачастую компенсирует (по крайней мере частично) дефектную копию. Таким образом Х-сцепленные заболевания обычно поражают мужчин в большей степени, чем женщин, поскольку у мужчин имеется только одна Х-хромосома. Х-сцепленные заболевания (подобно ШМТ-Х) не могут передаваться от отца к сыну Аутосомное — означает, что мутация локализована на хромосоме, отличной от Х или Y-хромосомы. Таким образом аутосомные заболевания поражают мужчин и женщин в равной степени. Аутосомно-рецессивное наследование — означает, что для проявления болезни требуется наличие двух копий дефектного гена. По одной копии унаследовано от каждого родителя, ни у кого из которых как правило нет проявлений заболевания. Аутосомно-доминантное наследование означает, что для проявления заболевания достаточно одной копии дефектного гена. Человек, унаследовавший дефектный ген от родителя, будет болен, как и этот родитель Когда ШМТ наследуется по аутосомно-доминантному типу, это легче проследить по родословной. Напротив, Х-сцепленные или аутосомно-рецессивные формы ШМТ могут возникать, казалось бы, ниоткуда. В действительности мать, или оба родителя, могут быть носителями, «тихим омутом» для генетических мутаций. Многие родители и не подозревают, что они носители, до тех пор, пока у них не родится ребенок с заболеванием ШМТ может также возникнуть, когда возникает новая мутация при зачатии ребенка. Такие мутации называются спонтанными, и после их возникновения они могут быть переданы следующим поколениям Ваш риск унаследовать или передать ШМТ в основном зависит от ее типа (см. «Каковы различные типы ШМТ») Хороший способ больше узнать о возможных рисках при планировании рождения детей — посоветоваться с лечащим врачом или генетическим консультантом. Так же можете посмотреть статью «Генетика и нервномышечные заболевания» ПОИСКИ МЕТОДОВ ЛЕЧЕНИЯ В 1991 году генетические основы ШМТ были совершенно неизвестны. Но уже через 10 лет ученые смогли идентифицировать 10 связанных с ШМТ генов. В настоящее время известно уже порядка 30 генов, мутации в которых вызывают ШМТ. Эти достижения позволяют проводить генетическое тестирование многих типов ШМТ, что существенно улучшает возможности диагностики. Не менее важным является то, что поиски новых генов, связанных с ШМТ, дают новые данные для разработки лечения, способного остановить или обратить заболевание. По мере обнаружения новых генов ученые исследуют, как и почему специфические генные мутации вызывают различные типы ШМТ. Эти данные, как полагают, помогут лучше предсказывать течение ШМТ у определенных индивидов и в конечном счете — лечить это заболевание Кроме того, ученые достигли существенного прогресса в понимании биологии аксонов и Шванновских клеток — клеток, производящих миелин в периферических нервах. Эти знания важны для того, чтобы научиться восстанавливать нормальное присутствие миелина. В то время как одни исследователи проводят лабораторные эксперименты, чтобы выяснить, сможет ли состав, именуемый «протеин теплового шока 90», использоваться в терапии ШМТ1А, другие проводят клинические исследования по изучению эффективности высоких доз аскорбиновой кислоты при этом же заболевании. miopatia.ru mioby.ru Перонеальная дистрофия Шарко-Мари-Тута (часть первая) : Генетические заболевания : Все про гены!Молекулярные причины возникновенияБолезнь Шарко-Мари-Тута вызванная мутациями, которые вызывают дефекты в белках нейронов. Нервные сигналы проводятся аксонами, которые покрыты миелиновой оболочкой. Большинство мутаций при ШМТ поражают миелиновую оболочку и некоторые аксоны. Наиболее распространенной причиной возникновения болезни (70-80% случаев) является дублирование большого региона в хромосоме 17p12, включающего в себя ген РМР22. Некоторые мутации поражают MFN2 ген, кодирующий деятельность митохондриального белка. Клетки содержат отдельные наборы генов в их ядре и митохондриях. В нервных клетках, митохондрии движутся вниз вдоль длинного аксона. При определенных формах ШМТ, мутировавший MFN2 ген вызывает образование большого кластера митохондрий или сгустка, который не может двигаться вниз по аксону к синапсам, что в свою очередь нарушает их функциональность. Выделяют следующие виды болезни: первичная демиелинизирующая нейропатия (ШМТ1, ШМТ3, и ШМТ4) и первичная аксональная нейропатия (ШМТ2), с частыми случаями наложения этих типов между собой. Другие клетки влияющие на появление болезни - это леммоциты (шванновские клетки), которые создают миелиновую оболочку, путем обертывания плазматической мембраны вокруг аксонов, эта структура, иногда сравнивается со швейцарским рулетом. Нейроны, шванновские клетки и фибробласты работая совместно образуют здоровый (работающий) нерв. Шванновские клетки и нейроны проводят молекулярные сигналы, регулирующие многочисленные процессы в организме. Именно эти сигналы нарушены при болезни Шарко-Мари-Тута. Демиелинизация шванновских клеток вызывает нарушение структуры и функций аксонов. Которые могут стать причиной дегенерации или нарушение функциональности аксона. Миелиновая оболочка позволяет нервным клеткам проводить сигналы значительно быстрее. Если миелиновая оболочка повреждается, то скорость проведения нервных сигналов замедляется. Скорость сигналов может быть определена путем проведения электромиографии - очень распространенного неврологического исследования. Также, когда аксон поврежден это приводит к уменьшению биопотенциала мышц (CMAP). Симптомы и течение болезни могут варьироваться. В некоторых случаях нарушается дыхание кроме того пораженным может быть слух, зрение, шея и плечевые мышцы. Распространенным явлением является сколиоз. Не исключено повреждение вертлужной впадины. При болезни возможны желудочно-кишечные расстройства, трудности при жевании, глотании и речи (атрофия медиального края голосовой складки). Атрофия мышц может вызвать тремор. Как правило, беременность обостряет ШMT, равно как и сильное эмоциональное напряжение. Нейропатические боли часто являются симптомом ШMT, хотя, как и другие симптомы ее наличие и тяжесть меняется в каждом отдельном случае. У некоторых людей, боль может быть очень сильной и мешать в повседневной жизни. Однако не у всех пораженных болезнь сопровождается болью. Когда боль присутствует как симптом ШMT, ее характер такой же как при других периферических нейропатиях, таких как постгерпетическая невралгия и комплексный региональный болевой синдром, и др. Болезнь Шарко-Мари-Тута может быть диагностирована при наличии характерных симптомов и с помощью измерения скорости электромиографии, биопсии нерва, а также путем анализа ДНК. ДНК-тестирование может дать окончательный, достоверный диагноз, но не все генетические маркеры для ШMT на сегодня являются известными. Первым симптомом болезни есть слабость в голени и деформация стопы, которая проявляется тяжестью при тыльном сгибании ноги и лодыжки, молотообразными пальцами и высоким подъемом. Но сами по себе признаки не являются основанием для установления диагноза болезни, именно поэтому пациенты должны быть направлены к неврологу или реабилитологу (физиотерапевту). Чтобы оценить мышечную слабость невролог попросит пациента пройти на носочках или переместить часть ноги против силы сопротивления. С целью выявления уровня чувствительности невролог будет определять глубокие сухожильные рефлексы (те, которые определяются с помощью молоточка), а именно коленный рефлекс (который при болезни снижен или отсутствует). Врач также спросит о семейной историю заболевания через наследственный характер передачи ШМТ. Отсутствие семейного анамнеза не исключает наличие болезни Шарко-Мари-Тута, но это позволяет врачу исключить другие причины невропатии, такие как диабет или влияние определенных химических веществ или наркотиков. В 2010 году, болезнь ШMT была одним из первых заболеваний, для которого с помощью секвенирования генома пораженного человека была точно определена генетическая причина болезни. В гене были обнаружены две мутации, из каких именно мутация Sh4TC2 была названа причиной возникновения болезни. Затем исследователи сравнили геном пациента с геномами матери, отца и семи братьев и сестер больного с и без болезни. Мать и отец имели одну нормальную и одну мутированную копии этого гена, в связи с чем симптомы болезни были умеренными, или их вообще не было. У потомства, унаследовавшего две копии аномальных генов болезнь проявлялась в полном объеме. Начальная стоимость секвенирования генома пациента составляла около 50 тысяч, но исследователи подсчитали, что вскоре оно будет стоить менее $ 5000 и станет общедоступным. Типы По состоянию на начало 2010 года, было идентифицировано мутации в 39 генах, вызывающих появление ШMT. Болезнь сначала можно классифицировать по основным клиническим категориям, а затем по подтипам в соответствии с этими мутациями. Тип 1, в первую очередь, влияет на миелиновую оболочку и является или доминирующим или рецессивным Х-сцепленным. 2 тип в свою очередь влияет на аксон и является или доминирующим или рецессивным. Другие типы являются смешанными. Клинические категории vse-pro-geny.ru Наследственная моторно-сенсорная нейропатия (НМСН, болезнь Шарко-Мари-Тута) тип I, II - ДНК-диагностика
Болезнь Шарко-Мари-Тута (ШМТ), или невральная амиотрофия Шарко-Мари, известная и как наследственная моторно-сенсорная невропатия (НМСН), - обширная группа генетически гетерогенных заболеваний периферических нервов, характеризующаяся симптомами прогрессирующей полинейропатии с преимущественным поражением мышц дистальных отделов конечностей. НМСН являются не только самым частым среди наследственных заболеваний периферической нервной системы, но и одним из самых частых наследственных заболеваний человека. Частота всех форм НМСН варьирует от 10 до 40:100000 в различных популяциях. Клинико-генетическая гетерогенность невральной амиотрофии Шарко-Мари, явилась основанием для поиска локусов, сцепленных с данными заболеваниями. К настоящему времени картировано более 40 локусов, отвечающих за наследственные моторно-сенсорные нейропатии, идентифицировано более двадцати генов, мутации в которых приводят к развитию клинического фенотипа НМСН. Описаны все типы наследования НМСН: аутосомно-доминантный, аутосомно-рецессивный и Х-сцепленный. Наиболее часто встречается аутосомно-доминантное наследование. Первичное поражение нерва приводит к вторичной слабости и атрофии мышц. В наибольшей степени страдают толстые «быстрые» нервные волокна, покрытые миелиновой оболочкой («мякотные» волокна) – такие волокна иннервируют скелетные мышцы. Длинные волокна повреждаются сильнее, поэтому в первую очередь нарушается иннервация наиболее дистальных (удаленных) мышц, испытывающих большую физическую нагрузку – это мышцы стоп и голеней, в меньшей степени – мышцы кистей рук и предплечий. Поражение чувствительных нервов приводит к нарушению болевой, тактильной и температурной чувствительности в стопах, голенях и кистях рук. В среднем заболевание начинается в возрасте 10-20 лет. Первыми симптомами являются слабость в ногах, изменение походки (штампующая, «петушиная» походка, или «степпаж»), подворачивание голеней, иногда возникают несильные преходящие боли в нижней части ног. В дальнейшем прогрессируют слабость мышц, происходит атрофия мышц голеней, ноги приобретают вид «перевернутых бутылок», часто происходит деформация стоп (стопы приобретают высокий свод, затем формируется так называемая «полая» стопа), в процесс вовлекаются мышцы кистей рук и предплечий. При осмотре врачом-невропатологом выявляется снижение или утрата сухожильных рефлексов (ахилловых, карпорадиальных, реже - коленных), сенсорные нарушения. Все моторно-сенсорные нейропатии в настоящее время по электронейромиографическим (ЭНМГ) и морфологическим признакам принято разделять на три основных типа: 1) демиелинизирующий (НМСНI), характеризующийся снижением скорости проведения импульса (СПИ) по срединному нерву, 2) аксональный вариант (НМСНII), характеризующийся нормальной или несколько сниженной СПИ по срединному нерву, 3) промежуточный вариант (intermedia) со СПИ по срединному нерву от 25 до 45 м/с. Величина СПИ, равная 38 м/с, определяемая по двигательной компоненте срединного нерва, считается условной границей между НМСНI (СПИ<38м/с) и НМСНII (СПИ>38м/с). Таким образом, ЭНМГ исследование приобретает особый смысл для ДНК-диагностики, поскольку позволяет выделить наиболее оптимальный алгоритм генетического обследования для каждой семьи. Возраст начала заболевания, его тяжесть и прогрессирование зависят от типа нейропатии, но могут сильно варьировать даже в пределах одной семьи. Наиболее часто встречается форма болезни НМСНIА - от 50% до 70 % всех случаев НМСН 1 типа в различных популяциях. В 10% случаев выявляются Х-сцепленные формы НМСН, среди которых преобладает форма с доминантным типом наследования – НМСНIX, составляющая 90% от всех Х-сцепленных полинейропатий. Среди НМСН II типа наиболее часто встречается доминантная форма – НМСНIIA – в 33% всех случаев (табл. 1). Наследственные моторно-сенсорные нейропатии с аутосомно-рецессивным типом наследования сравнительно редки, но клинически не отличимы от НМСН с аутосомно-доминантным типом наследования. НМСН 4D(Lom), 4С, 4H и 4J являются одними из таких болезней. Примечательно, что в генах NDRG1 и Sh4TC2 локализованы частые мутации, характерные для цыган. В ООО «Центр Молекулярной Генетики» разработан и проводится поиск наиболее частых мутаций цыганского происхождения, ответственных за развитие НМСН 4D(Lom) (Arg148X) и 4C (Arg1109X). Так же в ООО «Центр Молекулярной Генетики» разработана система поиска повторяющихся мутаций в генах GDAP1 (Leu239Phe), Sh4TC2 (Arg954X и Arg659Cys), FIG4 (Ile41Thr) и FGD4 (Met298Arg и Met298Thr), ответственных за аутосомно-рецессивные типы НМСН. Таблица 1. Гены, ответственные за развитие различных форм НМСН. (Синим цветом выделены гены, анализ которых проводится в ООО "Центр Молекулярной Генетики")
В ООО «Центр Молекулярной Генетики» разработана и проводится диагностика НМСН I, II и промежуточного типов с аутосомно-доминантным (АД), аутосомно-рецессивным (АР) и Х-сцепленным наследованием. Нами разработан набор для регистрации дупликаций в локусе гена PMP22 при болезни НМСН 1А с использованием двух микросателлитных повторов методом ПЦР. Набор предназначен для использования в диагностических лабораториях молекулярно-генетического профиля. При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала. Публикации по теме раздела Наследственная моторно-сенсорная нейропатия (Шарко-Мари-Тута)www.dnalab.ru |
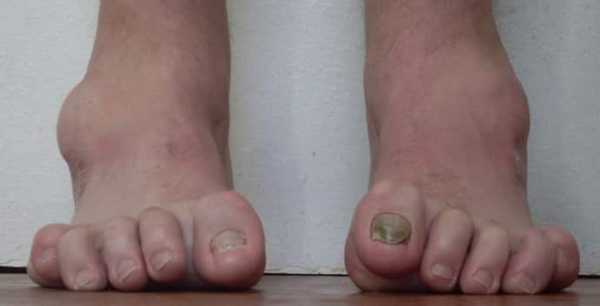












 Первое описание НМСН, известное в мировой литературе, было сделано французскими невропатологами Шарко и Мари в 1886 г., в статье «Относительно специфической формы прогрессирующей мышечной атрофии, часто семейной, начинающейся с поражения ступней и ног, и поздним поражением рук». Одновременно с ними заболевание описал Говард Тут в диссертации «Перонеальный тип прогрессирующей мышечной атрофии», который впервые сделал правильное предположение о связи заболевания с дефектами в периферических нервах. В России невропатолог, Давиденков Сергей Николаевич, впервые в 1934 году описал вариант невральной амиотрофии с усилением мышечной слабости при охлаждении.
Первое описание НМСН, известное в мировой литературе, было сделано французскими невропатологами Шарко и Мари в 1886 г., в статье «Относительно специфической формы прогрессирующей мышечной атрофии, часто семейной, начинающейся с поражения ступней и ног, и поздним поражением рук». Одновременно с ними заболевание описал Говард Тут в диссертации «Перонеальный тип прогрессирующей мышечной атрофии», который впервые сделал правильное предположение о связи заболевания с дефектами в периферических нервах. В России невропатолог, Давиденков Сергей Николаевич, впервые в 1934 году описал вариант невральной амиотрофии с усилением мышечной слабости при охлаждении.







.png){kind=link}