
|
656010, Алтайский край, город Барнаул,
ул.Эмилии Алексеевой, 53 корпус 1,39 корпус2
Телефон / Факс: +7 (3852) 22-62-77
[email protected]
|
Синдром ретта этоСиндром Ретта — ВикипедияМатериал из Википедии — свободной энциклопедии Идеограмма хромосомы человека X - Xq28. Красный локус обнаруживает ответственный ген в нижнем конце рисунка.Синдром Ретта (англ. Rett syndrome) — психоневрологическое наследственное заболевание, встречается почти исключительно у девочек с частотой 1:10000 — 1:15000, является причиной тяжёлой умственной отсталости у девочек[1]. Впервые болезнь была описана австрийским неврологом Андреасом Реттом (нем. Andreas Rett) в 1966 году. Развитие ребёнка до 6—18 месяцев протекает нормально, но потом у девочки начинают пропадать приобретённые речевые, двигательные и предметно-ролевые навыки. Характерным для данного состояния являются стереотипные, однообразные движения рук, их потирание, заламывание, при этом не носящие целенаправленного характера. Речь затрудняется, ответы становятся однообразными или эхолалическими, временами речь совсем пропадает (мутизм). Наблюдается низкий психологический тонус. Лицо ребёнка постепенно приобретает грустное, «неживое» выражение, взгляд становится расфокусированным или устремлённым в одну точку перед собой. Движения становятся заторможенными, но возможны приступы насильственного смеха вместе с приступами импульсивного поведения. Появляются судорожные припадки. Эти особенности напоминают поведение детей с ранним детским аутизмом. Несмотря на распространённую версию о том, что впервые синдром был описан в 1966 году, обнаружен он был в 1954 году, а всемирное признание как отдельное заболевание получил в 1983 году. В 1954 году Андреас Ретт обследовал двух девочек и отметил у них, кроме регресса психического развития, особые стереотипные движения в виде «сжимания рук» или напоминающие «мытьё рук». В своих записях он отыскал несколько подобных случаев, что натолкнуло его на мысль об уникальности заболевания. Отсняв на видеоплёнку своих пациенток, доктор Ретт отправился по всей Европе в поисках детей с похожими симптомами. В 1966 в Австрии он опубликовал свои исследования в паре немецких журналов, но они не получили всемирной огласки, даже после публикации на английском языке в 1977 году. Лишь в 1983 году, после публикации шведского исследователя Dr. Bengt Hagberg и его коллег[2], заболевание было выделено в отдельную нозологическую единицу и названо в честь своего первооткрывателя «синдромом Ретта». Было установлено, что синдром является генетическим заболеванием. По последним данным, к заболеванию приводят мутации в гене МЕСР2. Этот ген кодирует метил-СрG-связывающий белок 2(МеСР2) [3]. Если ген нормален, его белок в определённый момент развития мозга выключает из работы несколько других генов, и тогда мозг ребёнка развивается нормально. Если же ген поврежден мутацией, своевременное выключение не происходит. Мозг, пропустивший этот момент, начинает развиваться неправильно. Так возникает синдром Ретта. Ген МеСР2 находится в X-хромосоме. Поскольку у женщин таких хромосом две, женские плоды с синдромом Ретта обычно обладают одним нормальным и одним дефектным геном, что позволяет им дожить до рождения. У мужчин имеется только одна X-хромосома. Если она несёт дефектный ген МЕСР2, то у плода не остаётся нормальной копии гена, и он чаще всего погибает. Поэтому мальчики с синдромом Ретта рождаются очень редко. Несколько известных случаев болезни у мальчиков сопровождались синдромом Клайнфельтера с полисомией XXY, то есть одной лишней X-хромосомой. Это генетическое заболевание, состоящее из четырёх этапов, требующее реабилитации и ежедневной работы со специалистом по сохранению и приобретению новых навыков как моторных (ходьба), так и социопсихических. Синдром Ретта очень часто сопровождается судорогами (до 70 % случаев), нарушениями дыхания (гиповентиляция, гиперкапния, апноэ), сколиозом, желудочно-пищеводным рефлюксом, удлинённым интервалом QT на ЭКГ. Ряд больных достигает возраста 50 и более лет. Учёные из Эдинбургского университета под руководством Эдриана Берда сумели активировать ген МЕСР2 у лабораторных мышей. В результате симптомы синдрома Ретта у мышей исчезли[4]. Это позволяет надеяться на получение метода лечения синдрома и у людей. В настоящее время проводятся генетические и фармакологические исследования различных групп препаратов для лечения синдрома Ретта. Начиная с 1988 года проводятся мировые конгрессы по синдрому Ретта. В 2012 году конгресс проходил в Новом Орлеане, США. Очередной мировой конгресс состоялся в Казани 13-17 мая 2016 года. Это был первый мировой конгресс по редким болезням, который состоялся в России. Казань уже принимала Европейский конгресс по синдрому Ретта в 2011 году.
ru.wikipedia.org Синдром Ретта - причины, симптомы, диагностика и лечениеСиндром Ретта – генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника. Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного. Общие сведенияСиндром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году. Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера. Синдром Ретта Причины синдрома РеттаЭтиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается. Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани. При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей. Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК. В результате этого у больных синдромом Ретта наблюдается значительное превалирование процессов торможения в головном мозге, что отражается не только на физиологии центральной нервной системы, но и на ее морфологическом строении. Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко. Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца. Дефекты этого гена приводят и другим патологиям центральной нервной системы – варианту Запела, синдрому Луба (Х-сцепленная умственная отсталость у мальчиков), врожденной энцефалопатии. Некоторые исследователи относят эти состояния к атипичным формам синдрома Ретта. Симптомы синдрома РеттаУ новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям. Врачи-педиатры отмечают более медленный, нежели в норме, рост стоп и кистей в длину и замедление роста окружности головы. Иногда помимо неврологических проявлений может отмечаться нарушение работы печени, сердца, желудочно-кишечного тракта. Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе. Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения. Третья стадия синдрома Ретта называется псевдостационарной, так как при ней мало заметны признаки прогрессирования заболевания. Обычно она длится от 4 до 15 лет, состояние больных стабильно, однако наблюдаются судорожные приступы, глубокая умственная отсталость, гиперкинезы. В большинстве случаев синдрома Ретта этот этап оканчивается в пубертатном периоде. Четвертая стадия синдрома Ретта характеризуется уменьшением частоты эпилептических припадков вплоть до их исчезновения при прогрессировании двигательных расстройств. Большинство больных полностью теряют подвижность, возникает атрофия мышц, сосудистые нарушения в нижних конечностях, что может привести к развитию трофических язв. Из-за слабости мышечного корсета спины при синдроме Ретта возникает сколиоз или другие формы искривления позвоночника. Диагностика и лечение синдрома РеттаДиагностика синдрома Ретта производится на основании изучения анамнеза больного, его настоящего статуса, магнитно-резонансной томографии и энцефалографии, молекулярно-генетических анализов. Изучение наследственного анамнеза, как правило, не имеет особого смысла по причине спорадического характера мутаций гена MECP2. Характерными для синдрома Ретта являются нормальное развитие ребенка до 6-12 месяцев, возникновение мышечной гипотонии и беспокойства в раннем детстве, появление в дальнейшем атаксии и частых эпилептических припадков, стремительное утрачивание приобретенных навыков. В дальнейшем у больных регистрируется тяжелая умственная отсталость, мышечная слабость (вплоть до атрофии), искривление позвоночника, судорожные припадки. При осмотре больных синдромом Ретта выявляется отставание в росте и его остановка, резкое уменьшение окружности головы, отсутствие речи (на начальных этапах патологии характерна эхолалия). На магнитно-резонансной томографии головного мозга обнаруживается уменьшение размера органа, нечеткая дифференциация серого и белого вещества, базальных ганглиев, снижение складчатости коры больших полушарий. Электроэнцефалограмма подтверждает снижение фоновой активности головного мозга и резко ослабленную реакцию на внешние раздражители. Наиболее точную диагностическую информацию дают методы современной генетики – поиск делеций в локусе гена MECP2 или прямое секвенирование его последовательности для определения мутаций. Такое подтверждение синдрома Ретта возможно и в рамках пренатальной диагностики генетических заболеваний. Вспомогательную роль в установлении этого состояния может играть обследование внутренних органов (например, методами УЗИ) – у 20-30% больных выявляется недоразвитие печени или селезенки. Специфического лечения синдрома Ретта на сегодняшний день не существует. Имеются обнадеживающие данные некоторых исследовательских лабораторий, сотрудникам которых удалось «включить» ген MECP2 у мышей и тем самым добиться исчезновения симптомов заболевания. В сфере практической медицины пока доступна только симптоматическая терапия, однако и она сопряжена с рядом трудностей – в частности, эпилептические припадки при этом заболевании плохо поддаются устранению противосудорожными средствами. Также для лечения синдрома Ретта применяют ноотропные препараты, нарушения сна корректируют снотворными препаратами из группы барбитуратов или мелатонином. Прогноз и профилактика синдрома РеттаПрогноз синдрома Ретта неблагоприятный, так как это заболевание неуклонно ведет к тяжелой умственной отсталости, а также к ряду двигательных и неврологических нарушений. Пациенты с этой патологией при соответствующем уходе и симптоматическом лечении способны доживать до 40-50 лет, однако риск внезапной смерти у них довольно высокий. Значительно ухудшает прогноз синдрома Ретта и снижает продолжительность жизни больных наличие пороков развития внутренних органов, что имеет место примерно в трети случаев. Основная причина летального исхода – дыхательная или полиорганная недостаточность и внезапная смерть, у взрослых больных также велик риск инсульта. Профилактика синдрома Ретта возможна только в виде пренатальной диагностики этого заболевания генетическими методами. При наличии дефектной формы гена MECP2 у мальчика нарушение формирования головного мозга и внутренних органов можно заметить при профилактических ультразвуковых исследованиях во время беременности. www.krasotaimedicina.ru причины, 18 симптомов, 4 этапа, 6 методов леченияИстории детей с синдромом Ретта будто скопированы: беременность, роды проходят без осложнений, по шкале Апгар ребенку ставят высокий балл. Как это положено, в 3 месяца дети начинают держать голову. Сделать неуверенные шаги, сказать первые слова некоторые дети успевают, но в дальнейшем навыки теряются катастрофически быстро. За насколько недель ребенок теряет способность общаться с окружающими. Что такое синдром РеттаСиндром Ретта представляет собой прогрессирующее нейродегенеративное заболевание, которое влияет на развитие мозга ребенка и познавательные способности. Со временем он может вызвать серьезные проблемы с речью и коммуникацией, отсутствием координации и контроля мышц, непроизвольными движениями рук и замедлением роста. Синдром был впервые открыт в 1966 году Андреасом Реттом, австрийским педиатром — неврологом. Синдром Ретта — редкое, но тяжелое расстройство головного мозга, которое поражает только одну из 12 000 девочек. Синдром Ретта почти всегда поражает девочек, хотя мальчики могут заболеть в очень редких случаях. Младенцы, рожденные с синдромом Ретта, развиваются как обычные дети в течение первых нескольких месяцев их жизни. Они соответствуют типичным этапам развития, таким как визуальный контакт, взаимодействие с родителям, захватывание предметов. Однако, от 6 до 18 месяцев дети с синдромом Ретта начинают терять ранее приобретенные навыки, теряют интерес к игре и часто становятся все более раздражительными. По мере роста у детей с синдромом Ретта начинают проявляться проблемы с коммуникацией и контролем движения мышц, что приводит к трудностям с движениями, таким, как ходьба. У них могут появляться проблемы с дыханием, кормлением и глотанием, а также у них могут быть судороги и нарушения сна. У большинства заболевших детей диагностируется интеллектуальная отсталость.  Современный подход к лечению синдрома Ретта фокусируется на симптоматическом лечении, улучшении движения и коммуникации, а также на поддержке пациентов и их семей. В настоящее время нет лекарств от этой болезни. Ученые всего мира занимаются изучением, исследованием патогенеза этой болезни, для того чтобы найти то самое заветное лекарство. По распространённости в мире, это заболевание среди редких с подобной симптоматикой стоит сразу же после синдрома Дауна. В России по одним данным чуть больше сотни девочек с синдромом Ретта, по другим — 25 % от общего числа детей. Статистика хромает и вот ещё почему: синдром Ретта часто путают с детским церебральным параличом или аутизмом.Важно понимать, что хотя девочки и женщины, больные синдромом Ретта и испытывают трудности с общением, они имеют полный спектр эмоций и понимают больше, чем могут выразить. Причины синдромаСиндром вызван мутациями гена MECP2 на Х-хромосоме, одной из двух хромосом, определяющих пол человека. У девочек есть две Х-хромосомы, в то время как у мальчиков одна Х и одна У хромосома. Синдром Ретта чаще всего диагностируют у девочек, потому что у них есть вторая копия гена MECP2, которая способна работать правильно, и ребенок выживает. Из-за отсутствия у мальчиков копии гена MECP2, при его мутации в единственной Х-хромосоме такие дети либо погибают внутриутробно, либо очень сильно больны. В большинстве случаев синдром Ретта не наследуется (не передается от родителя к ребенку). Вероятность болезни у последующих детей у родителей, у которых есть один больной ребенок, составляет приблизительно 1%. До настоящего времени было идентифицировано более 200 мутаций гена MECP2, связанных с синдромом Ретта. При нормальном развитии человека ген MECP2 помогает создать белок (Methyl-CpG-связывающий белок 2), который регулирует активность других генов в организме. Когда уровень белка изменяется из-за дефектного гена, он заставляет другие гены в организме изменяться, что в конечном итоге влияет на нормальное развитие мозга. Дети с расстройством могут проявлять широкий спектр симптомов. На это может влиять местоположение, тип и тяжесть мутации гена, могут быть и другие факторы. Необходимы дополнительные исследования, чтобы лучше понять ген MECP2 и как он влияет на различные функции организма. СимптомыСимптомы могут сильно варьироваться. Хотя генетическое изменение, которое вызывает синдром Ретта, присутствует до рождения, в большинстве случаев ребенок с синдромом Ретта будет расти и нормально развиваться в течение первых 6-18 месяцев жизни до появления симптомов. Основные признакиСимптомы синдрома Ретта на начальном этапе могут включать следующие проявления.
 По мере того, как заболевшие дети растут, они могут потерять навыки, которые они уже узнали, такие как речь, способность использовать свои руки для таких действий, как прием пищи. Дети с синдромом Ретта могут проявлять меньший интерес к людям и предметам, которыми они пользовались. Большинство детей с расстройством начинают проявлять классические движения синдрома Ретта в возрасте от 1 до 4 лет. Они могут включать в себя сжатие, трение, скручивание, хлопание руками и повторяющиеся движения «рука в рот». По мере развития расстройства у детей будет все труднее контролировать свои мышцы, чтобы выполнять скоординированные двигательные движения. Это неврологическое состояние также называется апраксией или диспраксией. Некоторые дети с синдромом Ретта могут ходить и поддерживать эту способность, другие могут в конечном итоге потерять это умение, в то время как некоторые могут никогда не ходить самостоятельно. Дополнительные признакиДополнительные симптомы синдрома Ретта могут включать следующие признаки.
После первоначальной регрессии, развитие имеет тенденцию стабилизироваться у большинства девочек. Некоторые навыки (отсутствие интереса к общению) могут улучшаться, в то время как другие ( двигательные навыки) могут оставаться такими же или постепенно ухудшаться. Другие симптомы, такие как аномалии дыхания или судороги, могут возникать и уходить со временем. Несмотря на их многочисленные проблемы, у детей с синдромом Ретта есть своя уникальная личность, с симпатией и антипатией, они похожи на обычных детей. Этапы болезни
Диагностика синдрома РеттаСуществуют проблемы с ранней диагностикой, потому что не все врачи педиатры знакомы с первыми симптомами этого заболевания. Очень часто диагноз ставится поздно. Диагностика обычно начинается с выявления характерных симптомов болезни, а также сбора истории болезни и объективного осмотра. Диагностика может включать следующие процедуры.
В зависимости от симптомов, обнаруженных у ребенка, могут потребоваться дополнительные исследования для выявления любых проблем с приемом пищи и глотанием, желудочно-кишечных заболеваний или других медицинских проблем, которые могут возникнуть. Лечение синдрома РеттаВ настоящее время нет никакого конкретного лечения синдрома Ретта. Лечение детей с расстройством является сложным и разнообразным и должно быть настроено для удовлетворения конкретных потребностей ребенка. Лечение детей с синдромом Ретта часто включает следующие процедуры.
 Для лечения определенных симптомов, таких как судороги, назначается противоэпилептическая терапия. Дети с синдромом Ретта также подвержены повышенному риску развития сколиоза и сердечной аритмии — оба проявления могут потребовать дополнительного лечения. Для контроля сердечного ритма назначаются бета-блокаторы или применяется кардиостимулятор. Если сколиоз становится выраженным, используется корсет и спинальная хирургия, чтобы предотвратить дальнейшее развитие искривления позвоночника Выяснилось, что терапевтическая верховая езда, плавание, дельфинотерапия, гидротерапия и музыкальная терапия благотворно влияют на течение болезни. Важно постоянное выполнение физических нагрузок, занятия с логопедом, психологом, дефектологом. Если с ребенком не выполнять физических упражнений, не заниматься массажем, ЛФК, то он становиться лежачим и быстро теряет все приобретенные навыки. ПрогнозПерспективы для детей с синдромом Ретта варьируют, и во многом зависят от прогрессирования и тяжести симптомов. Хотя дети с синдромом Ретта нуждаются в помощи в большинстве повседневных дел, многие могут приобретать некоторые самостоятельные навыки, такие как прием пищи или хождение в туалет. Хотя общение, как правило, ограничено, многие девочки могут научиться общаться другими способами, например, используя дополнительные устройства связи. При оказании поддержки, люди с синдромом Ретта могут дожить до среднего возраста. Однако, из-за связанных с этим заболеванием состояний и осложнений для здоровья, люди с синдромом Ретта обычно имеют более короткую продолжительность жизни, чем в среднем у населения. Некоторые люди умирают в довольно молодом возрасте в результате осложнений, таких как аномалии сердечного ритма, пневмония и эпилепсия. Продолжительность жизни во многом зависит от реабилитации. Ранняя реабилитация имеет потенциал к улучшению состояния. kroha.info что это, причины, симптомы, диагностика, лечениеСиндром Ретта – врожденная патология головного мозга, характеризующаяся неполноценным развитием психики с нарушением интеллекта и приводящая к социальной дезадаптации. Это нервно-психическое генетическое заболевание обусловлено мутацией генов. Оно проявляется прогрессирующей умственной отсталостью преимущественно у девочек, гипотонусом мышц, шаткостью походки, сколиозом, запорами, утратой приобретенных навыков, дыхательными расстройствами, парезами и параличами. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой. Синдром у мальчиков встречается крайне редко. У них он несовместим с жизнью. Синдром был открыт в 1954 году психоневрологом из Австрии А. Реттом, благодаря которому и получил свое название. В отдельную нозологию он был выделен намного позже: примерно через 10 лет. Причины и патогенез патологии в настоящее время остаются неизвестными. Ученые считают, что спонтанная мутация генов Х-хромосомы — источник данной проблемы.
Внешние признаки патологии – маленькие руки и ноги, замедление темпов роста головы, признаки микроцефалии. Болезнь неуклонно прогрессирует и приводит к выраженной умственной отсталости, потере навыков самообслуживания и обездвиженности. Пациенты с синдромом Ретта склонны к желудочно-кишечным расстройствам и эпиприпадкам. дети с синдромом Ретта Диагностика заболевания заключается в проведении неврологического осмотра, МРТ головного мозга, ЭЭГ и молекулярно-генетического исследования. Синдром Ретта неизлечим. Больным показана симптоматическая терапия, целью которой является уменьшение выраженности отдельных проявлений недуга и облегчение общего состояния больного. ПричиныЭтиопатогенетические особенности синдрома Ретта являются достаточно сложными. Они обусловлены взаимодействием различных генов и их влиянием на головной мозг. Первопричина патологии — спонтанная мутация гена Х-хромосомы, приводящая к прекращению его экспрессии. Этот ген кодирует специфический белок, регулирующий транскрипцию определенных участков ДНК. При синдроме Ретта нарушается ингибирование генов, что приводит к неправильному формированию нервной ткани. Еще одна теория происхождения патологии — ломкость одного из участков короткого плеча Х-хромосомы. У больных нарушается ультраструктура нервных клеток: они уменьшаются в объеме, изменяется количество дендритов. Мозговые клетки не разрушаются, а начинают неправильно развиваться. Затрудненное образование нервной ткани никак не связано с процессом деструкции нейронов. Ученые отмечают, что при синдроме Ретта уменьшен размер головного мозга на 20% по сравнению с возрастной нормой. У пациентов преобладают процессы торможения в головном мозге. Это отражается на физиологии ЦНС, ее морфологическом строении и проявляется характерной клинической картиной. Если в геноме данный ген отсутствует полностью, плод погибает внутриутробно. Летальное состояние в основном характерно для мальчиков. У плода женского пола имеется две хромосомы: одна нормальная, а вторая дефектная. Это позволяет дожить ему до рождения. У мужчин же имеется только одна X-хромосома. Если она несет дефектный ген, то у плода не остается нормальной копии гена, и он чаще всего погибает. В нетипичных случаях синдром Ретта развивается у мальчиков и сопровождается менее яркой клинической картиной. В настоящее время к наследственной теории склоняется большинство ученых. Однако споры относительно этиопатогенетических факторов заболевания в ученом мире ведутся до сих пор. Менее известная теория развития патологии — метаболическая. Смысл ее заключается в том, что недуг возникает в результате нарушения обмена веществ, вызванного дисфункцией митохондрий. В настоящее время ни одна из этих теорий не доказана учеными. Они пока не смогли установить определенную закономерность, позволяющую утверждать стопроцентную взаимосвязь причины и следствия. Факторы, способствующие развитию патологии:
СимптомыПервые клинические признаки патологии возникают к концу первого года жизни. До года ребенок с синдромом Ретта растет и развивается обычными темпами.
Первые признаки недуга:
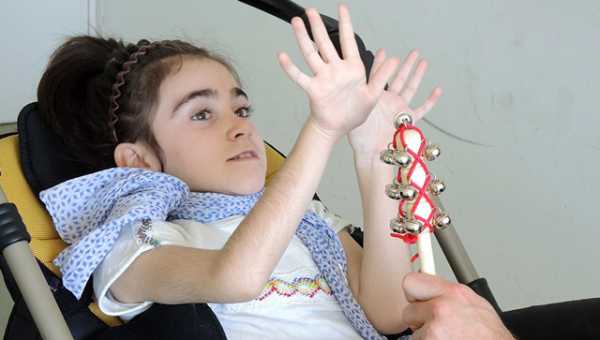
Больные дети отстают от сверстников в росте, имеют непропорциональные руки и ноги по отношению к туловищу, поздно начинают ходить, часто срыгивают, быстро и поверхностно дышат, страдают эпизодами внезапной остановки дыхания и судорожными припадками, имеют задержку моторики и речи. У больных детей возникают специфические движения рук, пропадают навыки удерживания предметов, появляются монотонные движения – перебирание пальцев или хлопки на уровне груди, сжимание кулачков, сплетение пальцев, поднесение рук к лицу, засовывание их в рот, покусывания рук или удары ими по разным частям тела. Движения рук полностью не исчезают, они становятся необычными. Ментальное развитие характеризуется умственной отсталостью и отсутствием познавательной деятельности, быстрой потерей приобретенных навыков. У больных развивается микроцефалия, появляются судорожные припадки, формируется сколиоз, обусловленный дистонией мышц спины. Непропорциональность головы по отношению к остальным частям тела становится очень заметной ближе к году. Больные дети асоциальны. Они не реагируют на любые внешние раздражители и похожи своим поведением на больных аутизмом. Гипервозбудимость, приступообразный плач, абсолютная беспомощность — характерные признаки патологии. Дети с синдромом Ретта могут подолгу раскачиваться из стороны в сторону и переминаться с ноги на ногу. У некоторых отмечается высокой болевой порог. Они проявляют агрессию по отношению к себе и окружающим: бьют близких и себя, кусают ногти, царапаются. К четырехлетнему возрасту рост мозга у больных детей останавливается полностью. Это приводит к дисфункции вегетативной нервной системы. У ребенка замедляется рост скелета, атрофируются мышцы, перестают нормально работать внутренние органы.
Эпиприпадки, бессонница и деменция с возрастом сменяются паркинсонизмом, стойкими нарушениями работы сердца и сосудов, потерей волос. У больных часто наблюдается дисморфизм лица, придающий им особый внешний вид. Женщины с синдромом Ретта фертильны, то есть способны к зачатию ребёнка. Прогрессирование заболевания характеризуется определенной стадийностью:
Все вышеперечисленные симптомы позволяют определить, на какой именно стадии находится больной с синдромом Ретта. Признаки заболевания могут варьироваться в зависимости от скорости прогрессирования болезни и некоторых индивидуальных особенностей организма. ДиагностикаДиагностика патологии основывается на данных анамнеза жизни больного и его неврологического статуса, результатах ядерно-магнитного резонанса и электроэнцефалографии, а также молекулярно-генетического анализа.
Характерные признаки синдрома Ретта, позволяющие заподозрить данный недуг:
Специалисты измеряют окружность головы ребенка и детально опрашивают родителей. Визуальный осмотр позволяет обнаружить отставание больного в росте от своих сверстников, уменьшение окружности головы, отсутствие речи. Инструментальная диагностика:
ЛечениеСиндром Ретта — генетическое заболевание, которое абсолютно не поддается терапии. Современная медицина бессильна перед генетикой. Симптоматическое лечение, облегчающее жизнь больным, также сопряжено с рядом трудностей. Синдром Ретта в настоящее время лечат в специализированных реабилитационных центрах, которые имеются практически во всех крупных городах нашей страны. Специалисты центров проводят развивающие занятия, адаптируя маленьких пациентов к окружающему миру.
Медикаментозная терапия смягчает симптоматические проявления патологии и облегчает общее состояние. Больным назначают:
Лечебный рацион показан с целью предупреждения истощения больных. Им рекомендовано частое и дробное питание, употребление продуктов с высоким содержанием жиров, клетчатки и витаминов. Диету составляют индивидуально каждому больному для набора веса. Физиотерапевтические процедуры, лечебный массаж, ЛФК — обязательные компоненты сложного лечебного процесса. Такие занятия способствуют развитию конечностей ребенка, повышают их гибкость, стимулируют мышечный тонус. Массаж и гимнастика поддерживают в исправном состоянии опорно-двигательный аппарат и противостоят дальнейшей деградации. Лечение музыкой активно применяется при синдроме Ретта. Такая терапия благотворно воздействует на организм, успокаивает и стимулирует интерес к окружающему миру. Работа с дефектологом, психологом, остеопатом помогает ребенку улучшить навыки коммуникации и моторику. Остеопатия положительно влияет на состояние позвоночника. Мануальная терапия обычно сочетается с другими методами реабилитации. Хороший эффект дают занятия со специально обученными животными, гидрореабилитация, арт-терапия. В настоящее время проводятся генетические и фармакологические исследования различных групп препаратов для лечения синдрома Ретта. Прогнозы
В настоящее время ученые разрабатывают специальные стволовые клетки, которых помогут побороть страшный недуг. Их вживление в организм позволит успешно противостоять прогрессированию болезни. Современные ученые экспериментально доказали, что синдром Ретта и его признаки вполне обратимы. Если восстановить функции гена, то можно устранить неврологические нарушения, которые провоцирует данная мутация. Эти исследования дают родителям и медикам большую надежду на восстановление большинства утраченных функций.
Поскольку синдром Ретта является генетическим заболеванием, предупредить его развитие невозможно. Профилактика патологии заключается в проведении ультразвукового скрининга всем беременным. Супружеские пары, имеющие в анамнезе хромосомные расстройства, с профилактической целью получают консультацию у генетика. Генетические тесты в процессе вынашивания ребенка позволяют выявить отклонение и принять решение о целесообразности прерывания беременности. Считается, что нарушения генотипа человека провоцируют негативные факторы — сложная экологическая обстановка и вредные привычки. Специалисты дают общие рекомендации своим пациентам: вести здоровый образ жизни, правильно питаться, заниматься спортом и следить за своим самочувствием. Женщинам в период беременности также необходимо следить за адекватным протеканием гестации и регулярно посещать акушера-гинеколога. Синдром Ретта – редкое заболевание, которое преимущественно диагностируется у девочек. Его невозможно победить окончательно. Современная медицина предлагает различные лекарственные препараты для уменьшения симптоматики недуга. Родителям не стоит отчаиваться, а следует помнить, что больной ребенок нуждается в бережном отношении, в любви и поддержке. Выполняя весь комплекс врачебных рекомендаций, можно значительно облегчить состояние малыша и улучшить его жизнь. Видео: примеры детей с синдромом Реттаsindrom.info Синдром Ретта у детей, что это такое, фото, симптомы и признакиЧто такое Синдром Ретта?Синдром Ретта – является наследственным заболеванием, психоневрологического генеза. Развивается в основном у девочек и приводит к развитию умственной отсталости. Данный синдром впервые описал невролог из Австрии Андреас Ретт. Особенности патологии заключаются в том, что ребенок до 18 месяцев развивается хорошо, но затем у ребенка начинают исчезать приобретенные, раннее, навыки: нарушается речь, двигательная активность, предметно-ролевая игра. Появляются однообразные движения рук, не преследующие какой-либо цели. Нередко ребенок их потирает и заламывает. Андреасом Реттом были обследованы две девочки, у которых он отметил регрессивный тип психоневрологического расстройства, проявляющийся движениями рук, напоминающих мытье. Причины возникновения синдромаПричины возникновения продолжают изучаться. Особая роль отводится генетической предрасположенности, а также мутации в геноме, связанного с X хромосомой. Мутация в гене, кодирующего метил-СрG-связывающий гликопротеин. приводит к нарушению правильного развития деятельности головного мозга в эмбриональном периоде развития. Если ген не изменен, то мозг и его отделы развиваются без патологии. Если произошла мутация, происходит поражение X- хромосомы. Так как у женщин их две, то одна X-хромосома – нормальная, а вторая – дефектная. С одной нормальной X-хромосомой девочкам удается появится на свет. Так как у мальчиков набор хромосом состоит из X и Y- хромосом, то их рождение с синдромом Ретта теоретически невозможна. Они погибают во внутриутробном периоде. Но существуют исключения. При полисомии XXY хромосомах, рождение мальчиков возможно (синдром Клайнфельтера). Симптомы синдром Ретта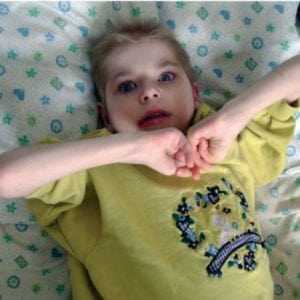 Больная девочка с синдромом Ретта Больная девочка с синдромом Ретта 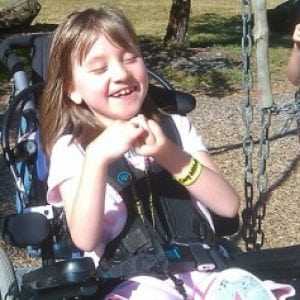   После рождения ребенок ничем не отличатся от своих сверстников в течение первых месяцев своей жизни. Врачи не могут заподозрить симптомы и выявить патологические изменения, характерные для синдрома Ретта. Окружность головки соответствует норме. Единственным признаком патологии может быть снижение и вялость мышечного тонуса. Также может изменяться температура тела (низкая, 35 градусов), бледные кожные покровы и высокая влажность ладоней. К 5-6 месяцам начинает выявляться отставание в физическом развитии ребенка. Он не ползает, не может переворачиваться на спинку или на бок. В большинстве случаев, в более старшем возрасте, ребенок не может удерживать свое тело в вертикальном положении и сидеть. Патогомоничные (отличительные) признаки синдрома Ретта:
Стадии болезниВыделяют 4 стадии заболевания:
Течение заболевания основывается на скорейшем установлении диагноза. Чем раньше был установлен диагноз, проведены лечебные и профилактические мероприятия, тем благоприятнее течение болезни. У разных детей течение патологии происходит по-разному. Если установление диагноза было выставлено в поздние сроки, велик риск развития тяжелейших осложнений, препятствующих нормальной жизни больного ребенка. ДиагностикаДиагностика заболевания основывается на сборе анамнеза (истории) заболевания, жизни больного, генетических изменений в гениалогическом древе, т.е наличия в семье/родственников данной патологии. Основным методом исследования является генетический анализ, который указывает на дефектную X-хромосому и с помощью данного анализа подтверждается болезнь Ретта. Проводят инструментальные методы исследования головного мозга:
При синдроме Ретта проводят дополнительные методы исследования, которые могут подтвердить или опровергнуть патологии других органов и систем на разных стадиях заболевания:
Постановка диагноза на начальных этапах основывается на клинических проявлениях заболевания. Чем сильнее выражены клинические признаки в возрасте не соответствующей стадии, тем короче продолжительность жизни ребенка. Дифференциальную диагностику синдрома Ретта у детей следует проводить с аутизмом. Что у синдрома Ретта что и у аутизма, есть одинаковые клинические проявления. Аутизм – дегенеративное расстройство нервной системы, развивающееся при нарушении формирования головного мозга и проявляющееся дефицитом общения, ограничением интересов и стереотипно повторяющимися действиями.
Лечение
Лечение синдрома Ретта основано на симптоматическом лечении. Устранить причину развития болезни и дальнейшее ее прогрессирования невозможно, но можно его замедлить. Лечение синдрома Ретта включает в себя: медикаментозную терапию, профилактическую и лечебно-физическую культуру.
 Мелатонин МелатонинПрофилактические мероприятия направлены на устранение венозного застоя в сосудах малого и большого кругов кровообращения, предупреждения развития острой сердечной недостаточности, развития отека легких, инсульта, субарахноидальных кровоизлияний.
Реабилитационные мероприятия при синдроме Ретта.
Прием лекарственных средств необходимо сочетать с лечебной физической культурой (ЛФК) и другими методами реабилитации. ЛФК позволяет улучшить осанку ребенка и предупредить развитие искривления позвоночника или прогрессирования сколиоза. С помощью ЛФК можно научить ребенка передвегаться. ЛФК необходимо сочетать с приемами мануального терапевта и лечебным массажем. Наиболее эффективна при синдроме Ретта музыкотерапия по схеме Томатиса. Специально созданная музыка, прослушиваемая детьми, ведет к улучшению психоэмоциональных показателей ребенка. Они начинают вступать в контакт с детьми и взрослыми. Положительной стороной является то, что музыкотерапию можно проводить в домашних условиях. Для того, чтобы ребенок адаптировался к окружающей среде можно использовать:
Также необходимы консультации логопедов, психологов, которые способны помочь ребенку «обрести себя», идти на контакты со взрослыми, выходить из депрессивного синдрома. ПитаниеПри данной патологии должно быть соблюдено лечебное питание. Диета с повышенным содержанием жиров + витамины и клетчатка. Необходимо кормить ребенка высококалорийными продуктами и специальными веществами для увеличения массы тела. Прием пищи должен быть каждые 3 часа. В случае недоедания развивается кахексия (резкое снижение веса). Прогноз жизниСколько живут дети с синдромом Ретта? Продолжительность жизни таких детей при своевременном установлении диагноза, правильной медикаментозной и профилактической терапии довольна высока. Девочки доживают до 30-40 лет. Мальчики, к сожалению, в лучшем случае, до 1-2 лет. Дети умирают в 4 стадии, с развитием тяжелейших осложнений: отек мозга, сердечная недостаточность, некроз кишечника, патология пищеварительного тракта, дисфункциональные патологии структур головного мозга. Видеозаписи по темеtvojajbolit.ru причины, симптомы, признаки, лечение. Что характерно для синдрома Ретта?Синдром Ретта — это одна из разновидностей прогрессирующего дегенеративного заболевания, характеризующаяся поражением ЦНС. Это редкая генетически обусловленная патология, которая развивается преимущественно у девочек в раннем возрасте. Ее можно отнести к группе психических и ментальных расстройств, поскольку недуг затрагивает жизненно важные отделы организма: функционирование головного мозга, нормальное развитие опорно-двигательного аппарата, ЦНС. В настоящее время медики не могут предложить адекватного лечения этого заболевания, так как природа его возникновения заложена на молекулярно-клеточном уровне. Чем отличается эта патология от всем известного аутизма? Каковы ее характерные симптомы? Можно ли вылечить заболевание с помощью приема лекарственных препаратов? Ответы на эти и многие другие вопросы можно узнать ознакомившись со статьей. Немного статистикиСиндром Ретта — это патология генетической природы, которая предположительно имеет наследственный характер. Для того чтобы более точно исследовать причины развития заболевания, ученые на протяжении нескольких лет проводили территориальный анализ распространения недуга, благодаря которому удалось выявить особую частоту случаев синдрома у детей из одного малонаселенного пункта. Эти «очаги» были отмечены в Норвегии, Венгрии и Италии. Заболевание начали активно изучать только на протяжении последних 15 лет. В настоящее время истинная природа этого недуга остается невыявленной до конца. Ученые со всего мира продолжают исследования в этом вопросе, главной целью которых считается поиск универсального биологического маркера. По их мнению, именно он впоследствии позволит создавать новые эффективные методы терапии не только этого синдрома, но и многих других заболеваний сходной генетической природы. Согласно статистическим данным, на 10-15 тысяч детей на свет появляется всего один ребенок женского пола с таким диагнозом. Синдром Ретта у мальчиков встречается крайне редко и несовместим с жизнью.  История возникновения заболеванияВ 1954 году известный австрийский педиатр Андреас Ретт впервые обнаружил симптомы этой патологии. Во время обследования двух девочек с явными расстройствами ментального спектра врач обратил внимание на нехарактерные для слабоумия признаки: заламывание рук, сжатие и долговременное сцепление пальцев, потирание кистей таким образом, будто ребенок моет их под водой. Такие движения повторялись с некоторой периодичностью и при этом сопровождались нестабильным эмоциональным состоянием, начиная полной погруженностью в себя и заканчивая резкими приступами криков. Впоследствии педиатр принялся за изучение этих двух случаев и обнаружил схожую клиническую картину в других историях болезней, что позволило ему выделить патологию в отдельную единицу. К 1966 году специалист диагностировал синдром еще у 31 девочки, а позже опубликовал результаты многолетних исследований в нескольких немецкоязычных изданиях. Однако на тот момент новая патология не получила широкой огласки, только спустя 20 лет она была признана на международном уровне и получила название «синдром Ретта» в честь первооткрывателя. С того времени ученые со всего мира принялись за активное изучение заболевания и его этиологии. Основные причиныКак только патологию вынесли в отдельное заболевание, специалисты начали выдвигать различные теории причин ее развития. Первоначально предположили, что недуг отличается генетической природой, то есть во всем виновата мутация генов. Отклонения такого характера объясняются присутствием большого числа кровных связей в родословной человека. С другой стороны, высказывались предположения о хромосомных аномалиях в качестве основной причины заболевания. Здесь речь идет о наличии ломкого участка в коротком плече Х-хромосомы. Ученые предполагают, что именно эта зона ответственна за формирование патологии. Проведенные впоследствии исследования на эту тему доказали, что у пациентов с таким диагнозом на самом деле имеются некоторые нарушения в хромосомах. Действительно ли этот фактор является главной причиной психических отклонений, до сих пор остается неизвестно. Единственное, что удалось установить точно, это возраст больных. По мнению врачей, первичные нарушения в работе мозга возникают с самого рождения ребенка, а уже к четвертому его году жизни развитие полностью прекращается. Более того, такие дети не могут полноценно развиваться и в физическом смысле.  Первые признаки синдрома РеттаВ самые первые месяцы жизни новорожденный выглядит абсолютно здоровым, у врачей обычно не возникает подозрений о каких-либо нарушениях. Окружность головы также находится в пределах нормативных показателей. Единственное, что может указывать на болезнь, это незначительная вялость мышц и симптомы гипотонии. К последним относится низкая температура, бледность кожных покровов и чрезмерная потливость ладошек. Примерно к 4-5 месяцам становятся заметны признаки отставания в развитии некоторых двигательных навыков (ползание, переворачивание на спинку). Впоследствии детям с синдромом Ретта с трудом удается сидеть, а также стоять. Синдром Ретта: симптомы заболеванияОтдельно необходимо акцентировать внимание на ключевых симптомах, по которым диагностируется заболевание. В медицинской практике известны случаи, когда вследствие неверного толкования признаков недуга ставился неверный диагноз, что в результате приводило к быстрому летальному исходу. Что характерно для синдрома Ретта?
Стадии развития синдромаПрогрессируя, заболевание проходит четыре основные стадии, каждая из которых имеет характерную клиническую картину.
Все вышеперечисленные симптомы позволяют определить, на какой именно стадии находится синдром Ретта. Признаки заболевания могут варьироваться в зависимости от скорости прогрессирования болезни и некоторых индивидуальных особенностей организма.  Как распознать патологию?Диагностика заболевания определяется наблюдаемой клинической картиной. При появлении подозрения на этот недуг детей обычно отправляют на аппаратное обследование. Оно включает тестирование состояния головного мозга с помощью КТ, измерение его биоэлектрической активности посредством ЭЭГ и ультразвуковое исследование. Заболевание, особенно на первых стадиях развития, нередко путают с аутизмом. Однако есть ряд отличий, которые позволяют дифференцировать эти две абсолютно разные патологии. В первые несколько месяцев жизни у детей-аутистов уже возникают характерные признаки заболевания, что нельзя сказать о синдроме Ретта. При аутическом расстройстве малыши нередко прибегают к различным манипуляциям с окружающими их предметами и обладают своеобразной грациозностью. Синдром Ретта у детей чаще всего проявляется скованностью в движениях из-за двигательных и мышечных нарушений. Более того, он сопровождается длительными эпилептическими припадками, замедлением роста головы и трудностями с дыханием. Таким образом, своевременно проведенная дифференциальная диагностика позволяет точно определить основную форму недуга, а также назначить соответствующее лечение и реабилитационные мероприятия. 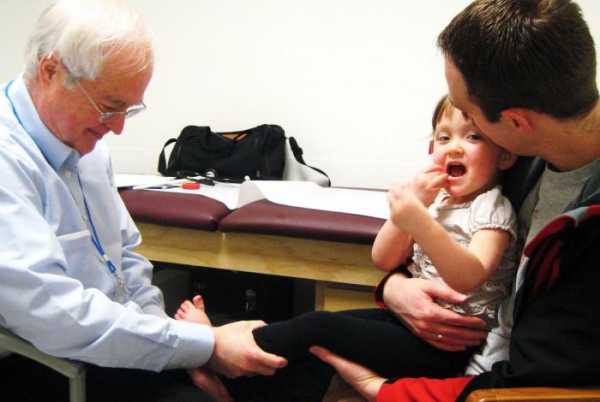 Медикаментозная терапияСовременные методы лечения заболевания, к сожалению, ограничены. Ключевое направление терапии — смягчение симптоматических проявлений патологии и облегчение состояния маленьких пациентов с помощью медикаментозных средств. Какие препараты назначают детям с диагнозом «синдром Ретта»? Лечение обычно включает:
Если эпилептические припадки повторяются с большой периодичностью, эффективность антиконвульсантов может оказаться невысокой. Как правило, детям с этим заболеванием назначают «Карбамазепин». Данное средство относится к категории сильных противосудорожных препаратов. Параллельно обычно прописывают «Ламотриджин». Данное средство относительно недавно появилось в фармакологии. Оно препятствует попаданию мононатриевой соли в ЦНС. Ученые обнаружили повышенное содержание этого вещества в спинномозговой жидкости у пациентов с диагнозом «синдром Ретта». Причины такого феномена до сих пор остаются неизвестны.  Традиционная терапияПомимо лекарственного лечения врачи рекомендуют специальную диету. Ее составляют в индивидуальном порядке для набора веса. Рацион должен быть насыщен клетчаткой, витаминами и калорийными продуктами. При этом требуется частое кормление (примерно каждые три часа). Такое питание способствует стабилизации состояния больных. Лечение также предполагает массаж и специальную гимнастику. Такие сеансы физических занятий способствуют развитию конечностей ребенка, повышают гибкость разных частей тела, а также стимулируют тонус мышц. Специалисты отмечают, что на детей с таким заболеванием благотворно воздействует музыка. Она не только успокаивает, но и стимулирует интерес к окружающему миру. Синдром Ретта у детей сегодня лечат в специализированных реабилитационных центрах. Они имеются практически во всех крупных городах. Здесь маленьких пациентов адаптируют к окружающему миру, для них проводят специальные развивающие занятия.  ПрогнозыДля назначения необходимого лечения ученые со всего мира продолжают активно изучать синдром Ретта. Прогноз специалистов в этом вопросе положительный. На данный момент разрабатывают специальные стволовые клетки, с помощью которых впоследствии можно будет побороть этот страшный недуг. Более того, такое «лекарство» уже было успешно опробовано на лабораторных мышах. В данной статье мы рассказали, что представляет собой синдром Ретта. Фото детей с таким недугом можно посмотреть в специализированных медицинских справочниках. Это очень редкое заболевание, которое преимущественно диагностируется у девочек. Если сравнивать с умственной отсталостью, этот синдром в первые несколько месяцев жизни ребенка не проявляется внешними признаками. В полгода возникают проблемы с психомоторным развитием, малыш теряет все навыки и совершенно перестает реагировать на окружающие явления и предметы. С течением времени клиническая картина ухудшается. К сожалению, это заболевание невозможно победить окончательно. Современная медицина предлагает различные лекарственные препараты для уменьшения симптоматики недуга. Кроме того, благотворно на здоровье ребенка влияют физиотерапевтические процедуры (гимнастика, массаж), занятия в специализированных реабилитационных центрах и медицинских учреждениях. Надеемся, что вся представленная в этой статье информация по теме окажется для вас на самом деле полезной. Будьте здоровы! fb.ru Синдром Ретта - это... Что такое Синдром Ретта?Синдром Ретта — психоневрологическое наследственное заболевание, встречается почти исключительно у девочек с частотой 1:10000 — 1:15000, является причиной тяжёлой умственной отсталости у девочек[1]. Впервые болезнь была описана австрийским неврологом Андреасом Реттом (нем. Andreas Rett) в 1966 году. Развитие ребёнка до 1—1,5 лет протекает нормально, но потом у девочки начинают распадаться только что приобретённые речевые, двигательные и предметно-ролевые навыки. Характерным для данного состояния являются стереотипные, однообразные движения рук, их потирание, заламывание, при этом не носящие целенаправленного характера. Речь затрудняется, ответы становятся однообразными или эхолалическими, временами речь совсем пропадает (мутизм). Наблюдается низкий психологический тонус. Лицо ребёнка постепенно приобретает грустное, «неживое» выражение, взгляд становится расфокусированным или устремлённым в одну точку перед собой. Движения становятся заторможенными, но возможны приступы насильственного смеха вместе с приступами импульсивного поведения. Появляются судорожные припадки. Эти особенности напоминают поведение детей с ранним детским аутизмом. ИсторияНесмотря на распространенную версию, что впервые синдром был описан в 1966 году, обнаружен он был в 1954 году, а всемирное признание, как отдельное заболевание получил в 1983 году. Так в 1954 году Андреас Ретт обследовал двух девочек и отметил у них кроме регресса психического развития, особые стереотипные движения в виде «сжимания рук» или напоминающие «мытье рук». В своих записях он отыскал несколько подобных случаев, что натолкнуло его на мысль об уникальности заболевания. Отсняв на видеопленку своих пациенток доктор Ретт отправился по всей Европе в поисках детей с похожими симптомами. В 1966 в Австрии он опубликовал свои исследования в паре немецких журналов, но они не получили всемирную огласку, даже после публикации на английском языке в 1977 году. Лишь в 1983 году после публикации шведского исследователя Dr. Bengt Hagberg и его коллег[2], заболевание было выделено в отдельную нозологическую единицу, и названо в честь его первооткрывателя «синдромом Ретта». Наследственная природаБыло установлено, что синдром имеет наследственную природу. Часто он наблюдается у монозиготных близнецов, у женщин связанных родственными связями по типу сёстры, мать-дочь, тётя-племянница, в популяционных изолятах[3]. По последним данным, к заболеванию приводят мутации в гене МЕСР2, находящемся в X-хромосоме. Этот ген кодирует метил-СрG-связывающий белок 2(МеСР2)[4]. Если ген нормален, его белок в определённый момент развития мозга выключает из работы несколько других генов, и тогда мозг ребёнка развивается нормально. Если же ген поражён мутацией, своевременное выключение не происходит. Мозг, пропустивший этот момент, начинает развиваться неправильно. Так возникает синдром Ретта. Ген МеСР2 находится в X-хромосоме. Поскольку у женщин этих хромосом две, женские эмбрионы с синдромом Ретта обычно обладают одним нормальным и одним дефектным геном, что позволяет им дожить до рождения. У мужчин же только одна X-хромосома. Если ген МеСР2 на этой хромосоме мутировал, у мужского эмбриона не остаётся нормальных генов, и эмбрион гибнет. Поэтому мальчики с синдромом Ретта рождаются очень редко. Несколько известных случаев болезни у мальчиков сопровождались синдромом Клайнфельтера с полисомией XXY, то есть одной лишней X хромосомой. Это наследственное заболевание не подлежит реабилитации и инвариантно заканчивается летальным исходом, обычно в возрасте 15-25 лет. Ряд больных достигает возраста 20-30 и более лет. Отчасти помогает коррекционная терапия на основе аминокислотных композитов и приём препаратов типа L-карнитина и кофермента Q в сочетании с витаминами групп A и B[5]. Новые данные свидетельствуют о возможности[6] применения креатина для реабилитации таких пациентов. Под влиянием креатина достоверно увеличивается метилирование ДНК, как генотипический маркер синдрома Ретта. Улучшения в познавательной и двигательной функций также наблюдались, хотя и не столь значимо. Учёные из Эдинбургского университета под руководством Эдриана Берда сумели активировать ген МЕСР2 у лабораторных мышей. В результате, симптомы синдрома Ретта у мышей исчезли[7]. Это позволяет надеяться на получение метода лечения синдрома и у людей. Примечания
Источники
Ссылкиdik.academic.ru |

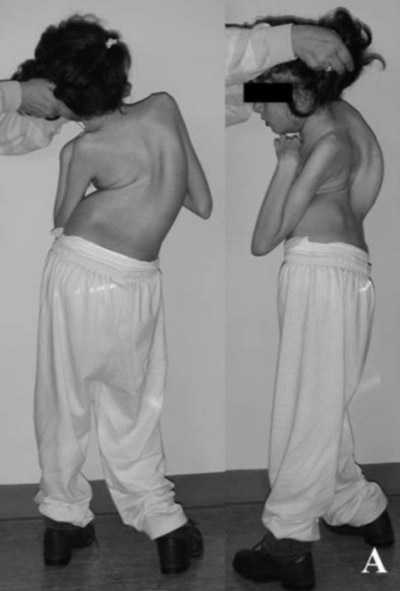 Четвертая стадия – уменьшение частоты эпиприпадков вплоть до их полного исчезновения. Больные теряют подвижность, возникает атрофия мышц, сосудистые нарушения в ногах, трофические язвы, различные формы искривления позвоночника. Возрастание тонуса мышц в отдельных конечностях приводит к образованию контрактур. У больных снижается масса тела вплоть до крайней степени истощения. Несмотря на существенные отклонения в физическом развитии, у пациентов отмечается полноценное половое созревание. Последняя стадия наблюдается у взрослых и длится до конца жизни пациента.
Четвертая стадия – уменьшение частоты эпиприпадков вплоть до их полного исчезновения. Больные теряют подвижность, возникает атрофия мышц, сосудистые нарушения в ногах, трофические язвы, различные формы искривления позвоночника. Возрастание тонуса мышц в отдельных конечностях приводит к образованию контрактур. У больных снижается масса тела вплоть до крайней степени истощения. Несмотря на существенные отклонения в физическом развитии, у пациентов отмечается полноценное половое созревание. Последняя стадия наблюдается у взрослых и длится до конца жизни пациента.












.png){kind=link}